RANKL逆シグナルによる骨吸収と骨形成の共役
本間 雅・池淵祐樹
(東京大学医学部附属病院 薬剤部)
email:本間 雅
DOI: 10.7875/first.author.2018.095
Coupling of bone resorption and formation by RANKL reverse signalling.
Yuki Ikebuchi, Shigeki Aoki, Masashi Honma, Madoka Hayashi, Yasutaka Sugamori, Masud Khan, Yoshiaki Kariya, Genki Kato, Yasuhiko Tabata, Josef M. Penninger, Nobuyuki Udagawa, Kazuhiro Aoki, Hiroshi Suzuki
Nature, 561, 195-200 (2018)
RANKLは骨芽細胞の系譜に発現し,破骨前駆細胞に発現するRANKを刺激して破骨細胞の分化および成熟を制御するタンパク質として知られている.近年の研究により,骨リモデリングにおける破骨細胞の成熟に関しては骨細胞に発現するRANKLが中心的な役割をはたすことが明らかにされたが,骨芽細胞に発現するRANKLの担う生理的な役割は不明確であった.この研究において,筆者らは,成熟の途上の破骨細胞から分泌された膜小胞に含まれるRANKが,骨芽細胞の表面のRANKLと結合してRANKL逆シグナルを活性化し,最終的に転写因子Runx2を活性化することにより骨形成を促進することを明らかにした.この逆シグナルにはRANKLの細胞内ドメインにあるプロリンリッチモチーフが必要であり,Pro29への点変異の導入により逆シグナルの活性化は抑制された.Pro29に点変異を導入したマウスにおいては骨吸収と骨形成の共役が抑制されており,骨芽細胞に発現するRANKLは小胞型RANKを認識する共役シグナルの受容タンパク質としてはたらくことが示唆された.
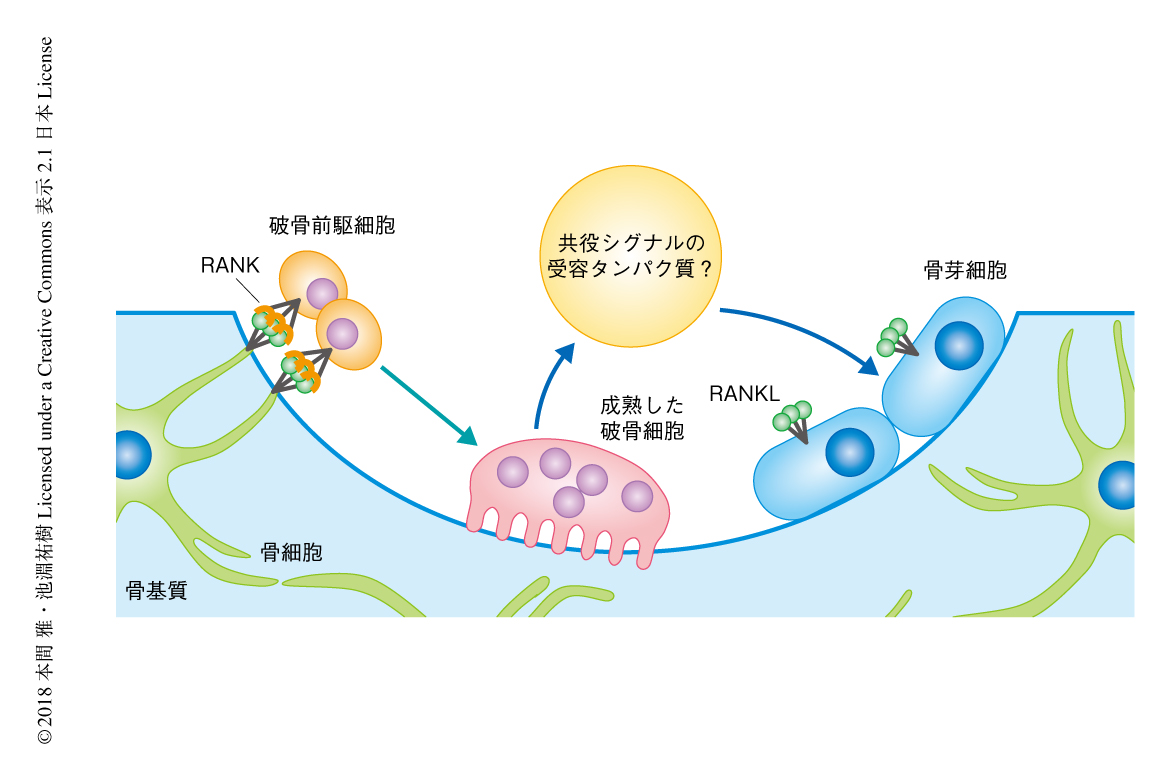
骨は破骨細胞による骨吸収と骨芽細胞による骨形成のサイクルである骨リモデリングをつうじ量および質が保たれている.骨吸収から骨形成への移行がスムーズに進行するためには,破骨細胞から骨芽細胞にむけたなんらかのシグナル伝達,すなわち,共役シグナルが必要と想定され,その分子機構の解明をめざした研究が精力的に進められている1).RANKLはTNFスーパーファミリーに属する膜貫通タンパク質で,骨芽細胞や骨細胞など骨芽細胞の系譜において発現が認められる.RANKLが破骨前駆細胞に発現するRANKと結合してシグナルを入力することにより破骨細胞の分化および成熟がひき起こされる.従来,骨の表面に局在する骨芽細胞が破骨細胞へのRANKLの入力を担うと考えられてきたが,骨細胞に選択的なRANKLノックアウトマウスを用いた解析を含む近年の研究の進展にともない,少なくとも,骨リモデリングの過程における破骨細胞の成熟については骨細胞がRANKLの主要な供給源であることが示された2).そのため逆に,骨芽細胞に発現するRANKLの生理的な役割は不明瞭な状況にあった(図1).
TNFスーパーファミリーには,細胞内にも逆シグナルを発生させる双方向性のシグナルタンパク質として機能する複数の例が知られている3).また,RANKLに結合する合成ペプチドが骨芽細胞の分化を促進する作用を示すという報告もある4).したがって,RANKLについても,骨芽細胞の分化を促進するシグナルの受容タンパク質として機能する可能性が想定された.もしこの仮説が正しいとすれば,シグナルを入力する役割を担うタンパク質としてはRANKか,あるいは,もうひとつのRANKL結合タンパク質として知られるオステオプロテグリンが候補となる.オステオプロテグリンは骨芽細胞の系譜に発現する分泌タンパク質であり,一般に,破骨細胞の分化を負に制御するタンパク質として知られている.しかしながら,オステオプロテグリンのノックアウトマウスにおいて骨芽細胞の骨形成の活性に変化は認められず5),RANKL逆シグナルのリガンドとして機能するとは考えにくかった.したがって,消去法的にRANKが候補となるが,新生骨の形成に寄与する骨芽細胞は吸収窩に位置する破骨細胞とは接触していないことが報告されている6).また,RANKLにより刺激された破骨前駆細胞の放出するSema4Dが骨芽細胞の吸収窩への移入をさまたげることも報告されている7)(新着論文レビュー でも掲載).これらをふまえると,破骨細胞の表面のRANKが骨芽細胞に対し直接的にRANKL逆シグナルを入力するとは考えにくかった.しかしながら,破骨細胞はその成熟の過程においてRANKを含む膜小胞を細胞の外に分泌することが報告されている8).そこで,筆者らは,この小胞型のRANKが骨芽細胞の表面のRANKLと結合してRANKL逆シグナルを入力するのではないかと仮説をたてた.
破骨細胞からのRANKの分泌について確認した.マウスの破骨前駆細胞様の細胞であるRAW264.7細胞を可溶型のRANKLの組換えタンパク質により刺激して成熟した破骨細胞の形成をひき起こし,成熟の過程のおのおのの段階において分泌物を分析したところ,RANKLによる刺激ののち60~120時間の,多核化が開始したのちの成熟の途上の破骨細胞の培養上清から超遠心により沈殿する画分にもっとも多くのRANKが検出され,無刺激あるいは多核化の開始のまえの前破骨細胞に由来する画分と比較して顕著に多かった.膜小胞のマーカータンパク質とRANKの共沈降は界面活性剤の処理により失われ,RANKが膜貫通タンパク質として膜小胞に含まれることが示唆された.
成熟の途上の破骨細胞に由来する膜小胞の骨芽細胞に対する作用について評価した.マウスの骨芽細胞様の細胞であるST2細胞を膜小胞により刺激し,骨芽細胞の分化のマーカータンパク質のmRNAのレベルでの発現を経時的に測定したところ,骨芽細胞の初期の分化のマスター転写因子であるRunx2およびその下流において制御される転写因子Osterixについて発現の上昇が認められ,時間の経過にともないI型コラーゲンおよびアルカリホスファターゼなど骨芽細胞の初期の分化のマーカータンパク質の発現の上昇も認められた.膜小胞を過剰量の可溶型RANKLにより前処理してRANKを被覆させた場合には,これらの発現は大幅に低下した.これらの結果から,小胞型RANKによる刺激が骨芽細胞においてRunx2の活性化をひき起こした可能性が示された.そこで,膜小胞に含まれるRANKの寄与を明確にするため,RANKの含有量が減少した成熟の途上の破骨細胞に由来する膜小胞の単離を試みた.CD40の細胞内ドメインをRANKの細胞内ドメインと置換したキメラタンパク質を,RANKの発現を抑制するshRNAとともにRAW264.7細胞に導入し,抗CD40抗体により刺激することで破骨細胞の成熟をひき起こした.この培養上清より精製した膜小胞においては,RANKの含有量が対照の約20%まで減少していた.この膜小胞によりST2細胞を刺激したところ,骨芽細胞の分化のマーカータンパク質の発現は大幅に抑制され,膜小胞に含まれるRANKが刺激の入力において中心的に関与することが示された.刺激をうけた骨芽細胞におけるRunx2の活性化についても検証した.成熟の途上の破骨細胞に由来する膜小胞により刺激したST2細胞から核タンパク質を抽出しイムノブロット法により解析したところ,核に局在するRunx2の量は刺激により増加することが確認された.
一連の結果をふまえ,小胞型RANKによる刺激が骨形成の促進につながるかどうか検証した.in vitroにおける検討として,ST2細胞を小胞型RANKの刺激のもと6日間にわたり培養し,そののち,12日間の追加の培養ののちvon Kossa染色したところ,骨形成の促進されているようすが観察された.そこで,in vivoにおける骨形成の促進の作用についても検証した.マウスの頭蓋骨に欠損を作製し小胞型RANKを含浸させたゼラチンヒドロゲルを留置したモデルを用い,4週間後の骨修復について評価した.その結果,膜小胞を含浸させたマウスにおいては新生骨の形成による欠損の修復が認められたが,対照のマウスにおいては欠損の修復は認められなかった.これらの結果から,破骨細胞の成熟の過程で分泌される小胞型RANKが骨芽細胞の初期の分化を促進し骨形成を促進する活性をもつことが示された.
骨芽細胞に発現するRANKLが小胞型RANKの受容タンパク質であることについて検証した.RANKLノックアウトマウスより初代骨芽細胞を単離し骨形成への影響について評価したところ,野生型のマウスに由来する骨芽細胞において観察された小胞型RANKによる骨形成の促進の作用は,RANKLノックアウトマウスに由来する細胞においてはほぼ完全に抑制された.骨芽細胞の分化のマーカータンパク質の発現についても同様であり,小胞型RANKにおける刺激の受容において骨芽細胞に発現するRANKLが必須であることが示された.
RANKLの下流においてRunx2の活性化に関与する細胞内シグナル伝達経路について解析した.mTOR複合体1がRunx2の活性化に関与することを示した既報にもとづき9),mTOR複合体1の阻害剤であるラパマイシンによる影響について評価したところ,小胞型RANKにより刺激したST2細胞におけるRunx2の核への移行に対し,ラパマイシンによる濃度に依存的な阻害が観察された.さらに,mTOR複合体1およびその上流の主要なタンパク質であるPI3KおよびAktについて,基質となるタンパク質のリン酸化を指標として活性の変化について評価したところ,小胞型RANKにより刺激したST2細胞において,PI3K-Akt-mTORC1シグナル伝達経路が活性化していることが明らかにされた.
これらシグナルタンパク質の活性化をひき起こす機構について解析を進めた.骨芽細胞に可溶型RANKあるいはオステオプロテグリンを添加するだけでは,RANKL逆シグナルの活性化は認められなかった.一方,小胞型RANKにより刺激した場合,膜小胞と細胞との接触部位にRANK-RANKL複合体が集積すると考えられ,このような細胞の表面におけるRANKLの集積がシグナルの入力に寄与する可能性が想定された.そこで,RANKLの細胞外ドメインを認識する単鎖化抗体のC末端側にイソロイシンジッパーを付加することで三量体化した組換えタンパク質を作製し,これによりST2細胞を刺激した.その結果,膜小胞による刺激と同様に,PI3K-Akt-mTORC1シグナル伝達経路の活性化が認められ,また,Runx2の核への移行も確認された.ラパマイシンの処理の影響,骨芽細胞の分化のマーカータンパク質の発現についても同様であった.これらの結果から,骨芽細胞の表面におけるRANKLの集積により細胞内に逆シグナルが生じることが示唆された.
Runx2は骨芽細胞の初期の分化を促進する一方で,後期の分化に対しては抑制的に機能することが知られている.そこで,RANKL逆シグナルについても同様の影響が認められるかどうか確認した.ST2細胞に対し,播種ののち1~6日のあいだにさきに述べた三量体化した抗RANKL抗体により刺激した場合,初期の分化が促進されるともに,培養12日目以降における後期の分化のマーカータンパク質の発現の時期も早まった.一方,播種ののち13~18日のあいだに刺激した場合,後期の分化のマーカータンパク質の発現は大幅に抑制された.同様に,初期の段階における刺激は骨形成を促進する一方,後期の段階における刺激は骨形成を抑制することも確認された.そこで,これらのRANKL逆シグナルの活性化の作用はRunx2を介するのかどうかを確認するため,Runx2のドミナントネガティブ変異体の影響について評価した.その結果,初期の分化の段階あるいは後期の分化の段階における刺激の作用は,刺激よりまえにRunx2のドミナントネガティブ変異体を導入することによりほぼ完全に抑制された.
ここまでの結果をふまえると,一連のシグナル伝達経路は生体において共役シグナルを媒介する可能性が想定されたことから,生体のレベルにおいて膜小胞の機能を抑制することを試みた.中性スフィンゴミエリナーゼに対する阻害剤GW4869が膜小胞の産生を抑制するという既報にもとづき10),in vitroにおいて小胞型RANKの分泌におよぼす影響について評価したところ,GW4869は成熟の途上の破骨細胞からの膜小胞の分泌を抑制したが,破骨細胞の成熟および骨吸収の活性におよぼす影響はわずかであった.そこで,外部から可溶型RANKLを投与することにより成熟した破骨細胞の過剰な形成を一過性にひき起こし,これに共役して生じる骨形成の促進を評価するマウスのモデルを用いて,GW4869の投与がおよぼす影響について評価した.その結果,GW4869は骨吸収と骨形成の共役による骨形成の促進を有意に抑制することが明らかにされた.
成熟の途上の破骨細胞に由来する膜小胞に選択性が高くかつ多量に含まれる膜タンパク質に対する抗体を投与することにより,生体において膜小胞の表面が被覆され,小胞型RANKと骨芽細胞の表面に局在するRANKLとの相互作用は立体障害により阻害されると想定した.成熟の途上の破骨細胞に由来する膜小胞をプロテオミクス法により解析した結果,標的となる膜タンパク質の候補としてIGSF8が同定された.IGSF8を認識する単鎖化抗体を三量体化した組換えタンパク質を作製し,小胞型RANKに対する中和活性をin vitroにおいて評価したところ,RANKL逆シグナルの入力の効率的な抑制が確認され,また,この抗IGSF8抗体は破骨細胞の成熟および骨吸収の活性に影響しなかった.そこで,さきのマウスのモデルを用いて同様に検討した結果,骨吸収と骨形成の共役による骨形成の促進が有意に抑制されることが明らかにされた.
RANKL逆シグナルの骨吸収と骨形成の共役への寄与について検証した.RANKLが双方向性のシグナルタンパク質である点,骨細胞は骨芽細胞から分化して生じる細胞である点などを考慮すると,骨芽細胞に特異的なRANKLノックアウトマウスの作出は困難であり,逆シグナルの受容能のみが選択的に抑制されたマウスのモデルが必要と考えられた.RANKLの細胞内ドメインには,一般にSH3ドメインと相互作用することの知られるプロリンリッチモチーフが存在する.SrcファミリーはSH3ドメインをもち,PI3Kを活性化するシグナルタンパク質である.そこで,Srcファミリーの阻害剤,あるいは,Srcのドミナントネガティブ変異体を導入した場合の影響について評価したところ,RANKL逆シグナルの活性化は強く抑制された.さらに,プロリンリッチモチーフのPro29あるいはPro39をAlaに置換したRANKL変異体を過剰に発現させた場合にも,RANKL逆シグナルの活性化は抑制された.これらの知見にもとづき,RANKLにPro29の点変異を導入したマウスを作出した.骨芽細胞におけるRANKLの発現および細胞の表面における存在量については,Pro29ホモ変異マウスと野生型のマウスのあいだでほとんど差は認められなかったが,Pro29ホモ変異マウスに由来する骨芽細胞においては,小胞型RANKにより刺激した際のPI3KおよびmTOR複合体1の活性化が抑制され,RANKL逆シグナルの受容能が低下していた.そこで,Pro29ホモ変異マウスおよび野生型のマウスに可溶型RANKLを投与し,骨吸収と骨形成の共役への影響について評価した.その結果,成熟した破骨細胞の一過性の過剰な形成について差異は認められなかったが,野生型のマウスに認められた骨形成の促進はPro29ホモ変異マウスにおいて大幅に抑制されており,RANKL逆シグナルが骨吸収と骨形成の共役に関与することが示唆された.
オステオプロテグリンのヘテロノックアウトマウスにおける代謝回転の速い骨表現型に対し,RANKLのPro29の点変異のおよぼす影響について評価した.オステオプロテグリンのヘテロノックアウトかつRANKLのPro29のホモ変異をもつマウス,および,オステオプロテグリンのヘテロ欠損かつRANKL野生型のマウスについて,腰椎における骨形態を計測した結果,成熟した破骨細胞の数について有意な差異は認められなかったが,RANKLのPro29ホモ変異マウスにおいて骨形成は有意に低下しており,RANKL逆シグナルは生理的な条件においても骨吸収と骨形成の共役に寄与すると考えられた.
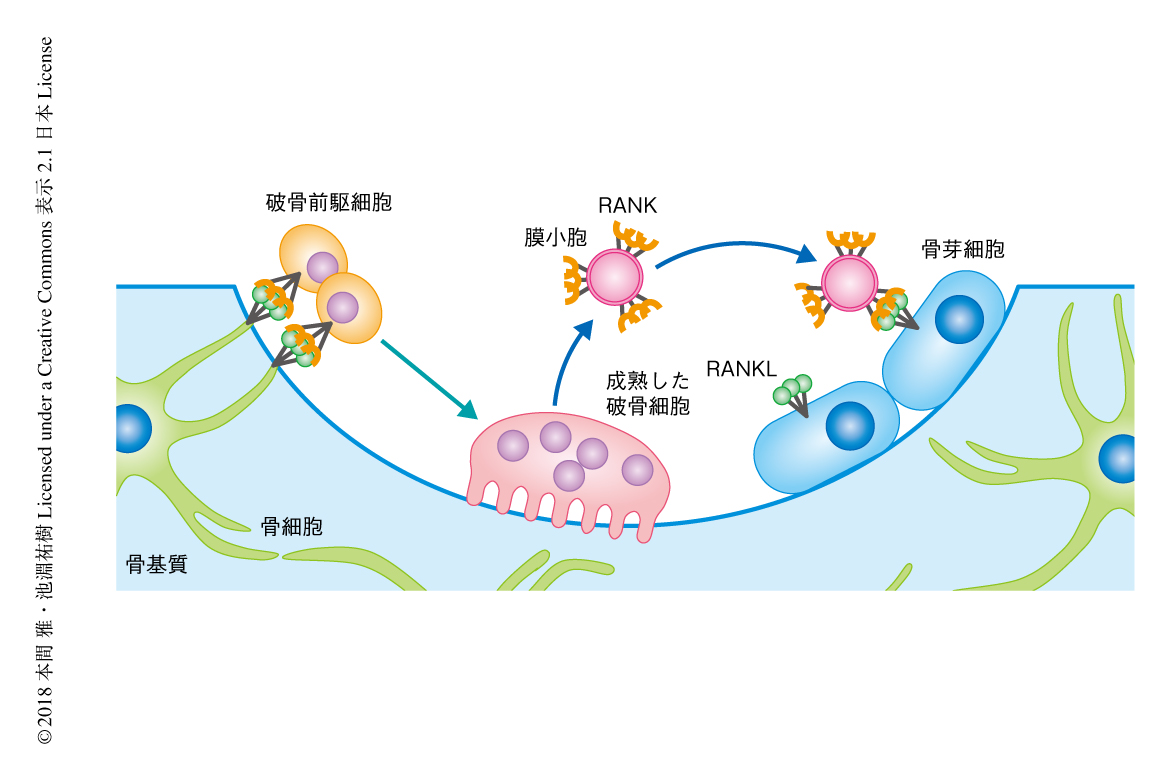
この研究において,筆者らは,骨芽細胞のRANKLは小胞型RANKを認識する共役シグナルの受容タンパク質としてはたらくことを明らかにした(図2).近年の研究により,RANKLの刺激をうけた直後の破骨前駆細胞より分泌されるSema4Dが骨芽細胞の分化を抑制し,骨吸収している破骨細胞から分泌されるCthrc1が骨芽細胞の後期の分化を促進することが示されている.そのため,この研究において見い出されたRANKL逆シグナルは,これらの共役シグナルの受容タンパク質と協調的に骨芽細胞の成熟および骨形成の開始のタイミングを精密に制御すると考えられる.今後,さらに多くの共役シグナルの受容タンパク質の関与およびその時空間的な制御の機構が明らかにされると期待される.
略歴:2014年 東京大学大学院薬学系研究科にて博士号取得,東京大学医学部附属病院 助教,同 特任准教授を経て,2017年より同 講師.
研究テーマ:慢性疾患の発症の機序および治療の標的の同定.
抱負:創薬に直接につながる研究を展開していきたい.
池淵 祐樹(Yuki Ikebuchi,)
東京大学医学部附属病院 助教.
© 2018 本間 雅・池淵祐樹 Licensed under CC 表示 2.1 日本
(東京大学医学部附属病院 薬剤部)
email:本間 雅
DOI: 10.7875/first.author.2018.095
Coupling of bone resorption and formation by RANKL reverse signalling.
Yuki Ikebuchi, Shigeki Aoki, Masashi Honma, Madoka Hayashi, Yasutaka Sugamori, Masud Khan, Yoshiaki Kariya, Genki Kato, Yasuhiko Tabata, Josef M. Penninger, Nobuyuki Udagawa, Kazuhiro Aoki, Hiroshi Suzuki
Nature, 561, 195-200 (2018)
この論文に出現する遺伝子・タンパク質のUniprot ID
RANKL(O35235), RANK(O35305), Runx2(Q08775), TNFスーパーファミリー, オステオプロテグリン(O08712), Sema4D(O09126), Osterix(Q8VI67), I型コラーゲン, アルカリホスファターゼ, CD40(P25942), mTOR複合体1, PI3K(Q6PF93), Akt, mTORC1, IGSF8(Q8R366), Srcファミリー, Src, Cthrc1(Q9D1D6)
要 約
RANKLは骨芽細胞の系譜に発現し,破骨前駆細胞に発現するRANKを刺激して破骨細胞の分化および成熟を制御するタンパク質として知られている.近年の研究により,骨リモデリングにおける破骨細胞の成熟に関しては骨細胞に発現するRANKLが中心的な役割をはたすことが明らかにされたが,骨芽細胞に発現するRANKLの担う生理的な役割は不明確であった.この研究において,筆者らは,成熟の途上の破骨細胞から分泌された膜小胞に含まれるRANKが,骨芽細胞の表面のRANKLと結合してRANKL逆シグナルを活性化し,最終的に転写因子Runx2を活性化することにより骨形成を促進することを明らかにした.この逆シグナルにはRANKLの細胞内ドメインにあるプロリンリッチモチーフが必要であり,Pro29への点変異の導入により逆シグナルの活性化は抑制された.Pro29に点変異を導入したマウスにおいては骨吸収と骨形成の共役が抑制されており,骨芽細胞に発現するRANKLは小胞型RANKを認識する共役シグナルの受容タンパク質としてはたらくことが示唆された.
はじめに
骨は破骨細胞による骨吸収と骨芽細胞による骨形成のサイクルである骨リモデリングをつうじ量および質が保たれている.骨吸収から骨形成への移行がスムーズに進行するためには,破骨細胞から骨芽細胞にむけたなんらかのシグナル伝達,すなわち,共役シグナルが必要と想定され,その分子機構の解明をめざした研究が精力的に進められている1).RANKLはTNFスーパーファミリーに属する膜貫通タンパク質で,骨芽細胞や骨細胞など骨芽細胞の系譜において発現が認められる.RANKLが破骨前駆細胞に発現するRANKと結合してシグナルを入力することにより破骨細胞の分化および成熟がひき起こされる.従来,骨の表面に局在する骨芽細胞が破骨細胞へのRANKLの入力を担うと考えられてきたが,骨細胞に選択的なRANKLノックアウトマウスを用いた解析を含む近年の研究の進展にともない,少なくとも,骨リモデリングの過程における破骨細胞の成熟については骨細胞がRANKLの主要な供給源であることが示された2).そのため逆に,骨芽細胞に発現するRANKLの生理的な役割は不明瞭な状況にあった(図1).
TNFスーパーファミリーには,細胞内にも逆シグナルを発生させる双方向性のシグナルタンパク質として機能する複数の例が知られている3).また,RANKLに結合する合成ペプチドが骨芽細胞の分化を促進する作用を示すという報告もある4).したがって,RANKLについても,骨芽細胞の分化を促進するシグナルの受容タンパク質として機能する可能性が想定された.もしこの仮説が正しいとすれば,シグナルを入力する役割を担うタンパク質としてはRANKか,あるいは,もうひとつのRANKL結合タンパク質として知られるオステオプロテグリンが候補となる.オステオプロテグリンは骨芽細胞の系譜に発現する分泌タンパク質であり,一般に,破骨細胞の分化を負に制御するタンパク質として知られている.しかしながら,オステオプロテグリンのノックアウトマウスにおいて骨芽細胞の骨形成の活性に変化は認められず5),RANKL逆シグナルのリガンドとして機能するとは考えにくかった.したがって,消去法的にRANKが候補となるが,新生骨の形成に寄与する骨芽細胞は吸収窩に位置する破骨細胞とは接触していないことが報告されている6).また,RANKLにより刺激された破骨前駆細胞の放出するSema4Dが骨芽細胞の吸収窩への移入をさまたげることも報告されている7)(新着論文レビュー でも掲載).これらをふまえると,破骨細胞の表面のRANKが骨芽細胞に対し直接的にRANKL逆シグナルを入力するとは考えにくかった.しかしながら,破骨細胞はその成熟の過程においてRANKを含む膜小胞を細胞の外に分泌することが報告されている8).そこで,筆者らは,この小胞型のRANKが骨芽細胞の表面のRANKLと結合してRANKL逆シグナルを入力するのではないかと仮説をたてた.
1.小胞型のRANKは骨芽細胞の分化を促進する
破骨細胞からのRANKの分泌について確認した.マウスの破骨前駆細胞様の細胞であるRAW264.7細胞を可溶型のRANKLの組換えタンパク質により刺激して成熟した破骨細胞の形成をひき起こし,成熟の過程のおのおのの段階において分泌物を分析したところ,RANKLによる刺激ののち60~120時間の,多核化が開始したのちの成熟の途上の破骨細胞の培養上清から超遠心により沈殿する画分にもっとも多くのRANKが検出され,無刺激あるいは多核化の開始のまえの前破骨細胞に由来する画分と比較して顕著に多かった.膜小胞のマーカータンパク質とRANKの共沈降は界面活性剤の処理により失われ,RANKが膜貫通タンパク質として膜小胞に含まれることが示唆された.
成熟の途上の破骨細胞に由来する膜小胞の骨芽細胞に対する作用について評価した.マウスの骨芽細胞様の細胞であるST2細胞を膜小胞により刺激し,骨芽細胞の分化のマーカータンパク質のmRNAのレベルでの発現を経時的に測定したところ,骨芽細胞の初期の分化のマスター転写因子であるRunx2およびその下流において制御される転写因子Osterixについて発現の上昇が認められ,時間の経過にともないI型コラーゲンおよびアルカリホスファターゼなど骨芽細胞の初期の分化のマーカータンパク質の発現の上昇も認められた.膜小胞を過剰量の可溶型RANKLにより前処理してRANKを被覆させた場合には,これらの発現は大幅に低下した.これらの結果から,小胞型RANKによる刺激が骨芽細胞においてRunx2の活性化をひき起こした可能性が示された.そこで,膜小胞に含まれるRANKの寄与を明確にするため,RANKの含有量が減少した成熟の途上の破骨細胞に由来する膜小胞の単離を試みた.CD40の細胞内ドメインをRANKの細胞内ドメインと置換したキメラタンパク質を,RANKの発現を抑制するshRNAとともにRAW264.7細胞に導入し,抗CD40抗体により刺激することで破骨細胞の成熟をひき起こした.この培養上清より精製した膜小胞においては,RANKの含有量が対照の約20%まで減少していた.この膜小胞によりST2細胞を刺激したところ,骨芽細胞の分化のマーカータンパク質の発現は大幅に抑制され,膜小胞に含まれるRANKが刺激の入力において中心的に関与することが示された.刺激をうけた骨芽細胞におけるRunx2の活性化についても検証した.成熟の途上の破骨細胞に由来する膜小胞により刺激したST2細胞から核タンパク質を抽出しイムノブロット法により解析したところ,核に局在するRunx2の量は刺激により増加することが確認された.
一連の結果をふまえ,小胞型RANKによる刺激が骨形成の促進につながるかどうか検証した.in vitroにおける検討として,ST2細胞を小胞型RANKの刺激のもと6日間にわたり培養し,そののち,12日間の追加の培養ののちvon Kossa染色したところ,骨形成の促進されているようすが観察された.そこで,in vivoにおける骨形成の促進の作用についても検証した.マウスの頭蓋骨に欠損を作製し小胞型RANKを含浸させたゼラチンヒドロゲルを留置したモデルを用い,4週間後の骨修復について評価した.その結果,膜小胞を含浸させたマウスにおいては新生骨の形成による欠損の修復が認められたが,対照のマウスにおいては欠損の修復は認められなかった.これらの結果から,破骨細胞の成熟の過程で分泌される小胞型RANKが骨芽細胞の初期の分化を促進し骨形成を促進する活性をもつことが示された.
2.細胞の表面におけるRANKLの集積が逆シグナルをひき起こす
骨芽細胞に発現するRANKLが小胞型RANKの受容タンパク質であることについて検証した.RANKLノックアウトマウスより初代骨芽細胞を単離し骨形成への影響について評価したところ,野生型のマウスに由来する骨芽細胞において観察された小胞型RANKによる骨形成の促進の作用は,RANKLノックアウトマウスに由来する細胞においてはほぼ完全に抑制された.骨芽細胞の分化のマーカータンパク質の発現についても同様であり,小胞型RANKにおける刺激の受容において骨芽細胞に発現するRANKLが必須であることが示された.
RANKLの下流においてRunx2の活性化に関与する細胞内シグナル伝達経路について解析した.mTOR複合体1がRunx2の活性化に関与することを示した既報にもとづき9),mTOR複合体1の阻害剤であるラパマイシンによる影響について評価したところ,小胞型RANKにより刺激したST2細胞におけるRunx2の核への移行に対し,ラパマイシンによる濃度に依存的な阻害が観察された.さらに,mTOR複合体1およびその上流の主要なタンパク質であるPI3KおよびAktについて,基質となるタンパク質のリン酸化を指標として活性の変化について評価したところ,小胞型RANKにより刺激したST2細胞において,PI3K-Akt-mTORC1シグナル伝達経路が活性化していることが明らかにされた.
これらシグナルタンパク質の活性化をひき起こす機構について解析を進めた.骨芽細胞に可溶型RANKあるいはオステオプロテグリンを添加するだけでは,RANKL逆シグナルの活性化は認められなかった.一方,小胞型RANKにより刺激した場合,膜小胞と細胞との接触部位にRANK-RANKL複合体が集積すると考えられ,このような細胞の表面におけるRANKLの集積がシグナルの入力に寄与する可能性が想定された.そこで,RANKLの細胞外ドメインを認識する単鎖化抗体のC末端側にイソロイシンジッパーを付加することで三量体化した組換えタンパク質を作製し,これによりST2細胞を刺激した.その結果,膜小胞による刺激と同様に,PI3K-Akt-mTORC1シグナル伝達経路の活性化が認められ,また,Runx2の核への移行も確認された.ラパマイシンの処理の影響,骨芽細胞の分化のマーカータンパク質の発現についても同様であった.これらの結果から,骨芽細胞の表面におけるRANKLの集積により細胞内に逆シグナルが生じることが示唆された.
3.RANKL逆シグナルはRunx2を介して骨芽細胞の分化に2相性の影響をおよぼす
Runx2は骨芽細胞の初期の分化を促進する一方で,後期の分化に対しては抑制的に機能することが知られている.そこで,RANKL逆シグナルについても同様の影響が認められるかどうか確認した.ST2細胞に対し,播種ののち1~6日のあいだにさきに述べた三量体化した抗RANKL抗体により刺激した場合,初期の分化が促進されるともに,培養12日目以降における後期の分化のマーカータンパク質の発現の時期も早まった.一方,播種ののち13~18日のあいだに刺激した場合,後期の分化のマーカータンパク質の発現は大幅に抑制された.同様に,初期の段階における刺激は骨形成を促進する一方,後期の段階における刺激は骨形成を抑制することも確認された.そこで,これらのRANKL逆シグナルの活性化の作用はRunx2を介するのかどうかを確認するため,Runx2のドミナントネガティブ変異体の影響について評価した.その結果,初期の分化の段階あるいは後期の分化の段階における刺激の作用は,刺激よりまえにRunx2のドミナントネガティブ変異体を導入することによりほぼ完全に抑制された.
4.骨芽細胞に発現するRANKLは共役シグナルを媒介する
ここまでの結果をふまえると,一連のシグナル伝達経路は生体において共役シグナルを媒介する可能性が想定されたことから,生体のレベルにおいて膜小胞の機能を抑制することを試みた.中性スフィンゴミエリナーゼに対する阻害剤GW4869が膜小胞の産生を抑制するという既報にもとづき10),in vitroにおいて小胞型RANKの分泌におよぼす影響について評価したところ,GW4869は成熟の途上の破骨細胞からの膜小胞の分泌を抑制したが,破骨細胞の成熟および骨吸収の活性におよぼす影響はわずかであった.そこで,外部から可溶型RANKLを投与することにより成熟した破骨細胞の過剰な形成を一過性にひき起こし,これに共役して生じる骨形成の促進を評価するマウスのモデルを用いて,GW4869の投与がおよぼす影響について評価した.その結果,GW4869は骨吸収と骨形成の共役による骨形成の促進を有意に抑制することが明らかにされた.
成熟の途上の破骨細胞に由来する膜小胞に選択性が高くかつ多量に含まれる膜タンパク質に対する抗体を投与することにより,生体において膜小胞の表面が被覆され,小胞型RANKと骨芽細胞の表面に局在するRANKLとの相互作用は立体障害により阻害されると想定した.成熟の途上の破骨細胞に由来する膜小胞をプロテオミクス法により解析した結果,標的となる膜タンパク質の候補としてIGSF8が同定された.IGSF8を認識する単鎖化抗体を三量体化した組換えタンパク質を作製し,小胞型RANKに対する中和活性をin vitroにおいて評価したところ,RANKL逆シグナルの入力の効率的な抑制が確認され,また,この抗IGSF8抗体は破骨細胞の成熟および骨吸収の活性に影響しなかった.そこで,さきのマウスのモデルを用いて同様に検討した結果,骨吸収と骨形成の共役による骨形成の促進が有意に抑制されることが明らかにされた.
RANKL逆シグナルの骨吸収と骨形成の共役への寄与について検証した.RANKLが双方向性のシグナルタンパク質である点,骨細胞は骨芽細胞から分化して生じる細胞である点などを考慮すると,骨芽細胞に特異的なRANKLノックアウトマウスの作出は困難であり,逆シグナルの受容能のみが選択的に抑制されたマウスのモデルが必要と考えられた.RANKLの細胞内ドメインには,一般にSH3ドメインと相互作用することの知られるプロリンリッチモチーフが存在する.SrcファミリーはSH3ドメインをもち,PI3Kを活性化するシグナルタンパク質である.そこで,Srcファミリーの阻害剤,あるいは,Srcのドミナントネガティブ変異体を導入した場合の影響について評価したところ,RANKL逆シグナルの活性化は強く抑制された.さらに,プロリンリッチモチーフのPro29あるいはPro39をAlaに置換したRANKL変異体を過剰に発現させた場合にも,RANKL逆シグナルの活性化は抑制された.これらの知見にもとづき,RANKLにPro29の点変異を導入したマウスを作出した.骨芽細胞におけるRANKLの発現および細胞の表面における存在量については,Pro29ホモ変異マウスと野生型のマウスのあいだでほとんど差は認められなかったが,Pro29ホモ変異マウスに由来する骨芽細胞においては,小胞型RANKにより刺激した際のPI3KおよびmTOR複合体1の活性化が抑制され,RANKL逆シグナルの受容能が低下していた.そこで,Pro29ホモ変異マウスおよび野生型のマウスに可溶型RANKLを投与し,骨吸収と骨形成の共役への影響について評価した.その結果,成熟した破骨細胞の一過性の過剰な形成について差異は認められなかったが,野生型のマウスに認められた骨形成の促進はPro29ホモ変異マウスにおいて大幅に抑制されており,RANKL逆シグナルが骨吸収と骨形成の共役に関与することが示唆された.
オステオプロテグリンのヘテロノックアウトマウスにおける代謝回転の速い骨表現型に対し,RANKLのPro29の点変異のおよぼす影響について評価した.オステオプロテグリンのヘテロノックアウトかつRANKLのPro29のホモ変異をもつマウス,および,オステオプロテグリンのヘテロ欠損かつRANKL野生型のマウスについて,腰椎における骨形態を計測した結果,成熟した破骨細胞の数について有意な差異は認められなかったが,RANKLのPro29ホモ変異マウスにおいて骨形成は有意に低下しており,RANKL逆シグナルは生理的な条件においても骨吸収と骨形成の共役に寄与すると考えられた.
おわりに
この研究において,筆者らは,骨芽細胞のRANKLは小胞型RANKを認識する共役シグナルの受容タンパク質としてはたらくことを明らかにした(図2).近年の研究により,RANKLの刺激をうけた直後の破骨前駆細胞より分泌されるSema4Dが骨芽細胞の分化を抑制し,骨吸収している破骨細胞から分泌されるCthrc1が骨芽細胞の後期の分化を促進することが示されている.そのため,この研究において見い出されたRANKL逆シグナルは,これらの共役シグナルの受容タンパク質と協調的に骨芽細胞の成熟および骨形成の開始のタイミングを精密に制御すると考えられる.今後,さらに多くの共役シグナルの受容タンパク質の関与およびその時空間的な制御の機構が明らかにされると期待される.
文 献
- Sims, N. A. & Martin, T. J.: Coupling the activities of bone formation and resorption: a multitude of signals within the basic multicellular unit. Bonekey Rep., 3, 481 (2014)[PubMed]
- Xiong, J., Onal, M., Jilka, R. L. et al.: Matrix-embedded cells control osteoclast formation. Nat. Med., 17, 1235-1241 (2011)[PubMed]
- Kisiswa, L., Osorio, C., Erice, C. et al.: TNFα reverse signaling promotes sympathetic axon growth and target innervation. Nat. Neurosci., 16, 865-873 (2013)[PubMed]
- Furuya, Y., Inagaki, A., Khan, M. et al.: Stimulation of bone formation in cortical bone of mice treated with a receptor activator of nuclear factor-κB ligand (RANKL)-binding peptide that possesses osteoclastogenesis inhibitory activity. J. Biol. Chem., 288, 5562-5571 (2013)[PubMed]
- Fei, Q., Guo, C., Xu, X. et al.: Osteogenic growth peptide enhances the proliferation of bone marrow mesenchymal stem cells from osteoprotegerin-deficient mice by CDK2/cyclin A. Acta. Biochim. Biophys. Sin., 42, 801-806 (2010)[PubMed]
- Andersen, T. L., Abdelgawad, M. E., Kristensen, H. B. et al.: Understanding coupling between bone resorption and formation: are reversal cells the missing link? Am. J. Pathol., 183, 235-246 (2013)[PubMed]
- Negishi-Koga, T., Shinohara, M., Komatsu, N. et al.: Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat. Med., 17, 1473-1480 (2011)[PubMed] [新着論文レビュー]
- Huynh, N., VonMoss, L., Smith, D. et al.: Characterization of regulatory extracellular vesicles from osteoclasts. J. Dent. Res., 95, 673-679 (2016)[PubMed]
- Singha, U. K., Jiang, Y., Yu, S. et al.: Rapamycin inhibits osteoblast proliferation and differentiation in MC3T3-E1 cells and primary mouse bone marrow stromal cells. J. Cell. Biochem., 103, 434-446 (2008)[PubMed]
- Trajkovic, K., Hsu, C., Chiantia, S. et al.: Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science, 319, 1244-1247 (2008)[PubMed]
活用したデータベースにかかわるキーワードと統合TVへのリンク
著者プロフィール
略歴:2014年 東京大学大学院薬学系研究科にて博士号取得,東京大学医学部附属病院 助教,同 特任准教授を経て,2017年より同 講師.
研究テーマ:慢性疾患の発症の機序および治療の標的の同定.
抱負:創薬に直接につながる研究を展開していきたい.
池淵 祐樹(Yuki Ikebuchi,)
東京大学医学部附属病院 助教.
© 2018 本間 雅・池淵祐樹 Licensed under CC 表示 2.1 日本
