AFF4遺伝子の機能獲得型の変異による先天異常症候群と転写伸長因子複合体およびコヒーシンによる遺伝子発現の制御機構
泉 幸佑・白髭克彦
(東京大学分子細胞生物学研究所 エピゲノム疾患研究センターゲノム情報解析研究分野)
email:泉 幸佑,白髭克彦
DOI: 10.7875/first.author.2015.037
Germline gain-of-function mutations in AFF4 cause a developmental syndrome functionally linking the super elongation complex and cohesin.
Kosuke Izumi, Ryuichiro Nakato, Zhe Zhang, Andrew C. Edmondson, Sarah Noon, Matthew C. Dulik, Ramakrishnan Rajagopalan, Charles P. Venditti, Karen Gripp, Joy Samanich, Elaine H. Zackai, Matthew A. Deardorff, Dinah Clark, Julian L. Allen, Dale Dorsett, Ziva Misulovin, Makiko Komata, Masashige Bando, Maninder Kaur, Yuki Katou, Katsuhiko Shirahige, Ian D. Krantz
Nature Genetics, 47, 338-344 (2015)
Cornelia de Lange症候群は精神および運動における発達の遅滞などを特徴とする多発奇形症候群のひとつであり,コヒーシン複合体やコヒーシン制御タンパク質をコードする遺伝子の変異により生じる.非典型的な症状を呈するCornelia de Lange症候群の患者から臨床所見の類似する患者を同定し,CHOPS症候群と名づけた.エクソーム塩基配列解析によりCHOPS症候群の患者の全例において,転写伸長因子複合体の構成タンパク質のひとつをコードするAFF4のミスセンス変異を同定した.そして,RNA-seq法およびChIP-seq法により,CHOPS症候群の患者とCornelia de Lange症候群の患者において,類似した遺伝子発現の異常,および,AFF4およびコヒーシンの構成タンパク質のゲノムへの局在の異常を見い出した.さらに,免疫沈降法およびウェスタンブロット法により,AFF4を含む転写伸長因子複合体,コヒーシンの構成タンパク質,RNAポリメラーゼIIのあいだの相互作用が検出された.以上から,CHOPS症候群およびCornelia de Lange症候群は転写伸長反応の異常による先天異常症候群であると考えられ,ヒトの初期発生における転写伸長反応の制御の重要性が明らかにされた.
Cornelia de Lange症候群を含む多くの先天異常症候群は,ヒトの初期発生の過程における遺伝子発現の異常により発症すると考えられている.近年,初期発生の過程における遺伝子発現の制御には,転写伸長反応の制御が重要な役割をはたすことが明らかにされてきた1).遺伝子の発現において,転写が開始しRNAポリメラーゼIIがmRNAを50塩基ほど転写したのち,RNAポリメラーゼIIは停止することが知られている.この停止したRNAポリメラーゼIIをさらなる転写伸長反応に移行させるためには,転写伸長因子複合体が必要である.転写伸長因子複合体はAFF4,ELL2,p-TEFb(CDK9/cyclinT1)などから構成される2).RNAポリメラーゼIIはC末端側に7残基からなるリピート配列をもち,このリピート配列がリン酸化されることにより転写開始反応および転写伸長反応が制御されている.転写開始の際にはリピート配列の5番目のSerがリン酸化され,転写伸長反応への移行の際には2番目のSerがリン酸化される3).転写伸長反応への移行に必要とされるリン酸化には,転写伸長因子複合体に存在するCDK9が必要である.この研究は,転写伸長因子複合体の構成タンパク質のひとつをコードするAFF4の変異による先天異常症候群を同定し,遺伝子発現の制御における転写伸長反応の制御の重要性について明らかにした.
なお,コヒーシンによる遺伝子発現の制御およびCornelia de Lange症候群については,武藤 彰彦, 領域融合レビュー, 4, e002 (2015) も参照されたい.
Cornelia de Lange症候群は精神および運動における発達の遅滞,低身長,小頭症,四肢の異常,先天性心疾患を含む内臓の奇形を特徴とする先天異常症候群のひとつである4).Cornelia de Lange症候群はコヒーシン複合体あるいはコヒーシン制御タンパク質をコードするNIPBL遺伝子,SMC1A遺伝子,SMC3遺伝子,RAD21遺伝子,HDAC8遺伝子の変異により発症する5-8).コヒーシンはSMC1A,SMC3,RAD21,STAG1あるいはSTAG2から構成され,環状の構造をもつ.コヒーシンはその環状の構造を利用してDNAをつつみこみ,細胞分裂における染色体分離において姉妹染色分体の早期の分離をふせぐ役割をはたす.コヒーシンの染色体への結合にはNIPBLやHDAC8といったコヒーシン制御タンパク質の必要なことが知られている.染色体分離の制御にくわえ,コヒーシンが転写制御にも役割をはたすことが知られてきているが,その機構にはいまだ不明な点が多い.
臨床診断として非典型的ではあるがCornelia de Lange症候群をうたがわれたがCornelia de Lange症候群の原因遺伝子に変異の認められない患者から,類似した臨床症状を示す3人の患者を同定した.共通する症状は,特徴的な顔貌,精神および運動における発達の遅滞,先天性の心疾患,肥満,慢性の肺疾患といった呼吸器の異常,低身長および骨形成の不全であり,これら症状の頭文字をとってCHOPS症候群と命名した.
CHOPS症候群の原因遺伝子を同定するためエクソーム塩基配列解析を行い,3人の患者に共通する遺伝子変異として,転写伸長因子複合体の構成タンパク質のひとつをコードするAFF4に3つのミスセンス変異を同定した.これらすべてのミスセンス変異は両親には認められず,精子あるいは卵子の形成の過程において生じた変異と考えられた.さらに,これらのミスセンス変異はAFF4のALF相同ドメインに集中していた.
ALF相同ドメインはAFF1にも保存されている.マウスにおいてはAff1遺伝子のALF相同ドメインにおけるミスセンス変異により小脳の失調および白内障といった表現型を呈することが知られ,その病態の分子機構として,ALF相同ドメインの変異によりユビキチンリガーゼであるSIAH1を介したプロテアソーム分解系が正常にはたらかなくなることが示されていた9).そこで,CHOPS症候群における病態の分子機構についても,AFF4の機能獲得型の変異によりSIAH1を介したプロテアソーム分解系がはたらかなくなるという可能性を考えた.
この仮説を検証するため,HEK293T細胞にAFF4およびSIAH1遺伝子の発現ベクターを強制発現させ,AFF4がSIAH1の機能により分解されるかどうかを調べた.野生型AFF4を発現させた際にはSIAH1遺伝子の共発現によりAFF4はすみやかに分解されたが,CHOPS症候群において同定された変異をもつAFF4を発現させた際にはSIAH1遺伝子を共発現させても変異AFF4はあまり分解されず,ミスセンス変異によりAFF4の安定性が増大している可能性が考えられた.CHOPS症候群の患者に由来する皮膚線維芽細胞をウェスタンブロット法により解析したところ,AFF4のタンパク質量は増加していた.これらの結果から,CHOPS症候群におけるAFF4の変異は機能獲得型の変異であることが示唆された.
CHOPS症候群の患者とCornelia de Lange症候群の患者には顔貌および発達遅滞といった共通する臨床症状が認められたため,類似した遺伝子発現の異常が存在する可能性を考え,CHOPS症候群の患者,Cornelia de Lange症候群の患者,健常人にそれぞれ由来する皮膚線維芽細胞を用いて,RNA-seq法により遺伝子の発現について検討した.興味深いことに,CHOPS症候群とCornelia de Lange症候群において類似した遺伝子発現パターンが認められ,CHOPS症候群とCornelia de Lange症候群とのあいだに類似した遺伝子発現の異常が存在することが明らかにされた.遺伝子発現の異常の原因を明らかにするため,抗AFF4抗体およびコヒーシン複合体の構成タンパク質のひとつであるRAD21に対する抗体を用いChIP-seq法により解析した.その結果,CHOPS症候群では発現の上昇している遺伝子において転写開始部位の近傍にAFF4およびRAD21の蓄積が認められた.そして,Cornelia de Lange症候群においても発現の上昇している遺伝子において転写開始部位の近傍にAFF4の蓄積が認められた.
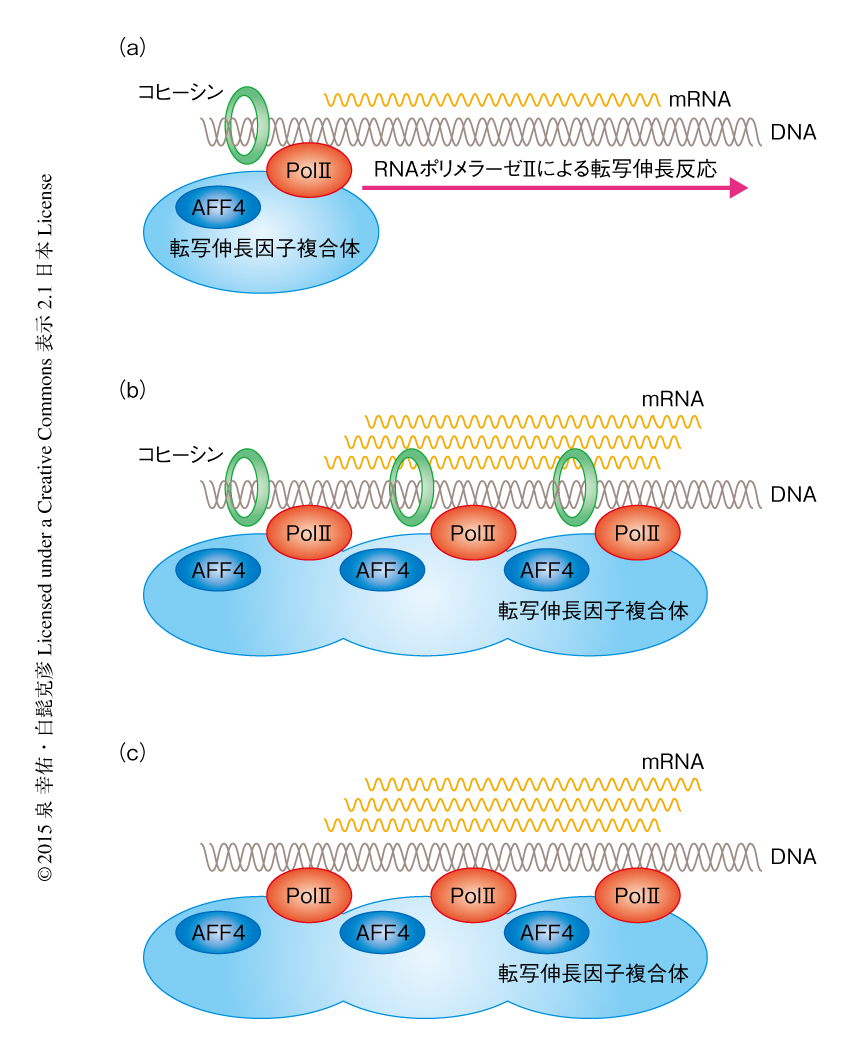
これらの結果から,コヒーシン,AFF4を含む転写伸長因子複合体,RNAポリメラーゼIIのあいだで相互作用のある可能性を考え,コヒーシンの構成タンパク質に対する抗体を用い免疫沈降法およびウェスタンブロット法により解析した.その結果,抗STAG1抗体により免疫沈降した場合には転写伸長因子複合体の構成タンパク質およびRNAポリメラーゼIIが共沈したが,抗STAG2抗体により免疫沈降した場合には転写伸長因子複合体の構成タンパク質あるいはRNAポリメラーゼIIは共沈しなかった.そのため,おもにSTAG1が転写伸長因子複合体を介した転写伸長反応の制御に関与している可能性が考えられた(図1).
近年,さまざまなクロマチン修飾タンパク質あるいはヒストン修飾タンパク質の異常による転写の異常とヒトの先天異常症候群との関連が明らかにされてきている.この研究においては,転写伸長因子複合体の構成タンパク質をコードするAFF4の生殖細胞系列における変異による遺伝病として,CHOPS症候群を報告した.CHOPS症候群は転写伸長反応の異常により発症する先天異常症候群であり,転写伸長反応がヒトの初期発生において重要なはたらきをしていることがあらためて証明されたといえる.さらに,CHOPS症候群とCornelia de Lange症候群における遺伝子発現パターンが類似していたことからヒントを得て,転写伸長因子複合体とコヒーシンとの相互作用を見い出した.そして,コヒーシンによる転写の制御の一部は転写伸長因子複合体とRNAポリメラーゼIIとの相互作用によることが明らかにされた.その相互作用はSTAG1によってのみ見い出されたことから,STAG1を含むコヒーシンとSTAG2を含むコヒーシンとに機能の違いがある可能性もうかびあがった.コヒーシンには姉妹染色分体の接着やDNA損傷の修復といったさまざまな機能が知られているが,そういったコヒーシンの多彩な役割がSTAG1を含むコヒーシンとSTAG2を含むコヒーシンによりどのように協調的に行われているかも,今後の課題のひとつと考えられる.
略歴:2008年 慶應義塾大学大学院医学研究科博士課程 修了,米国Rainbow Babies and Children’s Hospital,米国The Children’s Hospital of Philadelphiaを経て,2013年より東京大学分子細胞生物学研究所 助教.
研究テーマ:転写制御の異常,および,モザイク染色体異常症による先天異常症候群.
抱負:現在では治療がむずかしいと考えられている小児の先天異常症候群に対する治療法をみつけたい.
白髭 克彦(Katsuhiko Shirahige)
東京大学分子細胞生物学研究所 教授.
研究室URL:http://www.iam.u-tokyo.ac.jp/chromosomeinformatics/indexJ.html
© 2015 泉 幸佑・白髭克彦 Licensed under CC 表示 2.1 日本
(東京大学分子細胞生物学研究所 エピゲノム疾患研究センターゲノム情報解析研究分野)
email:泉 幸佑,白髭克彦
DOI: 10.7875/first.author.2015.037
Germline gain-of-function mutations in AFF4 cause a developmental syndrome functionally linking the super elongation complex and cohesin.
Kosuke Izumi, Ryuichiro Nakato, Zhe Zhang, Andrew C. Edmondson, Sarah Noon, Matthew C. Dulik, Ramakrishnan Rajagopalan, Charles P. Venditti, Karen Gripp, Joy Samanich, Elaine H. Zackai, Matthew A. Deardorff, Dinah Clark, Julian L. Allen, Dale Dorsett, Ziva Misulovin, Makiko Komata, Masashige Bando, Maninder Kaur, Yuki Katou, Katsuhiko Shirahige, Ian D. Krantz
Nature Genetics, 47, 338-344 (2015)
要 約
Cornelia de Lange症候群は精神および運動における発達の遅滞などを特徴とする多発奇形症候群のひとつであり,コヒーシン複合体やコヒーシン制御タンパク質をコードする遺伝子の変異により生じる.非典型的な症状を呈するCornelia de Lange症候群の患者から臨床所見の類似する患者を同定し,CHOPS症候群と名づけた.エクソーム塩基配列解析によりCHOPS症候群の患者の全例において,転写伸長因子複合体の構成タンパク質のひとつをコードするAFF4のミスセンス変異を同定した.そして,RNA-seq法およびChIP-seq法により,CHOPS症候群の患者とCornelia de Lange症候群の患者において,類似した遺伝子発現の異常,および,AFF4およびコヒーシンの構成タンパク質のゲノムへの局在の異常を見い出した.さらに,免疫沈降法およびウェスタンブロット法により,AFF4を含む転写伸長因子複合体,コヒーシンの構成タンパク質,RNAポリメラーゼIIのあいだの相互作用が検出された.以上から,CHOPS症候群およびCornelia de Lange症候群は転写伸長反応の異常による先天異常症候群であると考えられ,ヒトの初期発生における転写伸長反応の制御の重要性が明らかにされた.
はじめに
Cornelia de Lange症候群を含む多くの先天異常症候群は,ヒトの初期発生の過程における遺伝子発現の異常により発症すると考えられている.近年,初期発生の過程における遺伝子発現の制御には,転写伸長反応の制御が重要な役割をはたすことが明らかにされてきた1).遺伝子の発現において,転写が開始しRNAポリメラーゼIIがmRNAを50塩基ほど転写したのち,RNAポリメラーゼIIは停止することが知られている.この停止したRNAポリメラーゼIIをさらなる転写伸長反応に移行させるためには,転写伸長因子複合体が必要である.転写伸長因子複合体はAFF4,ELL2,p-TEFb(CDK9/cyclinT1)などから構成される2).RNAポリメラーゼIIはC末端側に7残基からなるリピート配列をもち,このリピート配列がリン酸化されることにより転写開始反応および転写伸長反応が制御されている.転写開始の際にはリピート配列の5番目のSerがリン酸化され,転写伸長反応への移行の際には2番目のSerがリン酸化される3).転写伸長反応への移行に必要とされるリン酸化には,転写伸長因子複合体に存在するCDK9が必要である.この研究は,転写伸長因子複合体の構成タンパク質のひとつをコードするAFF4の変異による先天異常症候群を同定し,遺伝子発現の制御における転写伸長反応の制御の重要性について明らかにした.
なお,コヒーシンによる遺伝子発現の制御およびCornelia de Lange症候群については,武藤 彰彦, 領域融合レビュー, 4, e002 (2015) も参照されたい.
1.AFF4の変異によるCHOPS症候群の同定
Cornelia de Lange症候群は精神および運動における発達の遅滞,低身長,小頭症,四肢の異常,先天性心疾患を含む内臓の奇形を特徴とする先天異常症候群のひとつである4).Cornelia de Lange症候群はコヒーシン複合体あるいはコヒーシン制御タンパク質をコードするNIPBL遺伝子,SMC1A遺伝子,SMC3遺伝子,RAD21遺伝子,HDAC8遺伝子の変異により発症する5-8).コヒーシンはSMC1A,SMC3,RAD21,STAG1あるいはSTAG2から構成され,環状の構造をもつ.コヒーシンはその環状の構造を利用してDNAをつつみこみ,細胞分裂における染色体分離において姉妹染色分体の早期の分離をふせぐ役割をはたす.コヒーシンの染色体への結合にはNIPBLやHDAC8といったコヒーシン制御タンパク質の必要なことが知られている.染色体分離の制御にくわえ,コヒーシンが転写制御にも役割をはたすことが知られてきているが,その機構にはいまだ不明な点が多い.
臨床診断として非典型的ではあるがCornelia de Lange症候群をうたがわれたがCornelia de Lange症候群の原因遺伝子に変異の認められない患者から,類似した臨床症状を示す3人の患者を同定した.共通する症状は,特徴的な顔貌,精神および運動における発達の遅滞,先天性の心疾患,肥満,慢性の肺疾患といった呼吸器の異常,低身長および骨形成の不全であり,これら症状の頭文字をとってCHOPS症候群と命名した.
CHOPS症候群の原因遺伝子を同定するためエクソーム塩基配列解析を行い,3人の患者に共通する遺伝子変異として,転写伸長因子複合体の構成タンパク質のひとつをコードするAFF4に3つのミスセンス変異を同定した.これらすべてのミスセンス変異は両親には認められず,精子あるいは卵子の形成の過程において生じた変異と考えられた.さらに,これらのミスセンス変異はAFF4のALF相同ドメインに集中していた.
2.CHOPS症候群における病態の分子機構
ALF相同ドメインはAFF1にも保存されている.マウスにおいてはAff1遺伝子のALF相同ドメインにおけるミスセンス変異により小脳の失調および白内障といった表現型を呈することが知られ,その病態の分子機構として,ALF相同ドメインの変異によりユビキチンリガーゼであるSIAH1を介したプロテアソーム分解系が正常にはたらかなくなることが示されていた9).そこで,CHOPS症候群における病態の分子機構についても,AFF4の機能獲得型の変異によりSIAH1を介したプロテアソーム分解系がはたらかなくなるという可能性を考えた.
この仮説を検証するため,HEK293T細胞にAFF4およびSIAH1遺伝子の発現ベクターを強制発現させ,AFF4がSIAH1の機能により分解されるかどうかを調べた.野生型AFF4を発現させた際にはSIAH1遺伝子の共発現によりAFF4はすみやかに分解されたが,CHOPS症候群において同定された変異をもつAFF4を発現させた際にはSIAH1遺伝子を共発現させても変異AFF4はあまり分解されず,ミスセンス変異によりAFF4の安定性が増大している可能性が考えられた.CHOPS症候群の患者に由来する皮膚線維芽細胞をウェスタンブロット法により解析したところ,AFF4のタンパク質量は増加していた.これらの結果から,CHOPS症候群におけるAFF4の変異は機能獲得型の変異であることが示唆された.
3.CHOPS症候群とCornelia de Lange症候群の類似した表現型
CHOPS症候群の患者とCornelia de Lange症候群の患者には顔貌および発達遅滞といった共通する臨床症状が認められたため,類似した遺伝子発現の異常が存在する可能性を考え,CHOPS症候群の患者,Cornelia de Lange症候群の患者,健常人にそれぞれ由来する皮膚線維芽細胞を用いて,RNA-seq法により遺伝子の発現について検討した.興味深いことに,CHOPS症候群とCornelia de Lange症候群において類似した遺伝子発現パターンが認められ,CHOPS症候群とCornelia de Lange症候群とのあいだに類似した遺伝子発現の異常が存在することが明らかにされた.遺伝子発現の異常の原因を明らかにするため,抗AFF4抗体およびコヒーシン複合体の構成タンパク質のひとつであるRAD21に対する抗体を用いChIP-seq法により解析した.その結果,CHOPS症候群では発現の上昇している遺伝子において転写開始部位の近傍にAFF4およびRAD21の蓄積が認められた.そして,Cornelia de Lange症候群においても発現の上昇している遺伝子において転写開始部位の近傍にAFF4の蓄積が認められた.
これらの結果から,コヒーシン,AFF4を含む転写伸長因子複合体,RNAポリメラーゼIIのあいだで相互作用のある可能性を考え,コヒーシンの構成タンパク質に対する抗体を用い免疫沈降法およびウェスタンブロット法により解析した.その結果,抗STAG1抗体により免疫沈降した場合には転写伸長因子複合体の構成タンパク質およびRNAポリメラーゼIIが共沈したが,抗STAG2抗体により免疫沈降した場合には転写伸長因子複合体の構成タンパク質あるいはRNAポリメラーゼIIは共沈しなかった.そのため,おもにSTAG1が転写伸長因子複合体を介した転写伸長反応の制御に関与している可能性が考えられた(図1).
おわりに
近年,さまざまなクロマチン修飾タンパク質あるいはヒストン修飾タンパク質の異常による転写の異常とヒトの先天異常症候群との関連が明らかにされてきている.この研究においては,転写伸長因子複合体の構成タンパク質をコードするAFF4の生殖細胞系列における変異による遺伝病として,CHOPS症候群を報告した.CHOPS症候群は転写伸長反応の異常により発症する先天異常症候群であり,転写伸長反応がヒトの初期発生において重要なはたらきをしていることがあらためて証明されたといえる.さらに,CHOPS症候群とCornelia de Lange症候群における遺伝子発現パターンが類似していたことからヒントを得て,転写伸長因子複合体とコヒーシンとの相互作用を見い出した.そして,コヒーシンによる転写の制御の一部は転写伸長因子複合体とRNAポリメラーゼIIとの相互作用によることが明らかにされた.その相互作用はSTAG1によってのみ見い出されたことから,STAG1を含むコヒーシンとSTAG2を含むコヒーシンとに機能の違いがある可能性もうかびあがった.コヒーシンには姉妹染色分体の接着やDNA損傷の修復といったさまざまな機能が知られているが,そういったコヒーシンの多彩な役割がSTAG1を含むコヒーシンとSTAG2を含むコヒーシンによりどのように協調的に行われているかも,今後の課題のひとつと考えられる.
文 献
- Lin, C., Garrett, A. S., De Kumar, B. et al.: Dynamic transcriptional events in embryonic stem cells mediated by the super elongation complex (SEC). Genes Dev., 25, 1486-1498 (2011)[PubMed]
- Lin, C., Smith, E. R., Takahashi, H. et al.: AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol. Cell, 37, 429-437 (2010)[PubMed]
- Heidemann, M., Hintermair, C., Voss, K. et al.: Dynamic phosphorylation patterns of RNA polymerase II CTD during transcription. Biochim. Biophys. Acta, 1829, 55-62 (2013)[PubMed]
- Liu, J. & Krantz, I. D.: Cornelia de Lange syndrome, cohesin, and beyond. Clin. Genet., 76, 303-314 (2009)[PubMed]
- Gillis, L. A., McCallum, J., Kaur, M. et al.: NIPBL mutational analysis in 120 individuals with Cornelia de Lange syndrome and evaluation of genotype-phenotype correlations. Am. J. Hum. Genet., 75, 610-623 (2004)[PubMed]
- Deardorff, M. A., Kaur, M., Yaeger, D. et al.: Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. Am. J. Hum. Genet., 80, 485-494 (2007)[PubMed]
- Deardorff, M. A., Wilde, J. J., Albrecht, M. et al.: RAD21 mutations cause a human cohesinopathy. Am. J. Hum. Genet., 90, 1014-1027 (2012)[PubMed]
- Deardorff, M. A., Bando, M., Nakato, R. et al.: HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature, 489, 313-317 (2012)[PubMed]
- Oliver, P. L., Bitoun, E., Clark, J. et al.: Mediation of Af4 protein function in the cerebellum by Siah proteins. Proc. Natl. Acad. Sci. USA, 101, 14901-14906 (2004)[PubMed]
著者プロフィール
略歴:2008年 慶應義塾大学大学院医学研究科博士課程 修了,米国Rainbow Babies and Children’s Hospital,米国The Children’s Hospital of Philadelphiaを経て,2013年より東京大学分子細胞生物学研究所 助教.
研究テーマ:転写制御の異常,および,モザイク染色体異常症による先天異常症候群.
抱負:現在では治療がむずかしいと考えられている小児の先天異常症候群に対する治療法をみつけたい.
白髭 克彦(Katsuhiko Shirahige)
東京大学分子細胞生物学研究所 教授.
研究室URL:http://www.iam.u-tokyo.ac.jp/chromosomeinformatics/indexJ.html
© 2015 泉 幸佑・白髭克彦 Licensed under CC 表示 2.1 日本
