脱ユビキチン化酵素USP8の機能獲得型変異はCushing病をひき起こす
駒田 雅之
(東京工業大学大学院生命理工学研究科 生体システム専攻情報生物学分野)
email:駒田雅之
DOI: 10.7875/first.author.2015.002
Mutations in the deubiquitinase gene USP8 cause Cushing's disease.
Martin Reincke, Silviu Sbiera, Akira Hayakawa, Marily Theodoropoulou, Andrea Osswald, Felix Beuschlein, Thomas Meitinger, Emi Mizuno-Yamasaki, Kohei Kawaguchi, Yasushi Saeki, Keiji Tanaka, Thomas Wieland, Elisabeth Graf, Wolfgang Saeger, Cristina L. Ronchi, Bruno Allolio, Michael Buchfelder, Tim M. Strom, Martin Fassnacht, Masayuki Komada
Nature Genetics, 47, 31-38 (2015)
Cushing病は脳下垂体の副腎皮質刺激ホルモン産生細胞の腺腫からの副腎皮質刺激ホルモンの過剰な分泌が原因となり発症する.10人のCushing病の患者から摘出した下垂体腫瘍の全エキソーム解析の結果,4人の腫瘍において脱ユビキチン化酵素USP8の遺伝子に体細胞変異が見い出された.これらの変異はUSP8の14-3-3結合モチーフに集中しており,変異を生じたUSP8は14-3-3タンパク質との結合能を失っていた.その結果,USP8変異体は14-3-3結合モチーフの直前で限定分解をうけやすくなり,限定分解により生じたC末端側の酵素活性ドメインのみからなる断片は高い脱ユビキチン化活性を示した.USP8はEGFにより活性化されたEGF受容体を脱ユビキチン化し,そのリソソームへの輸送および分解を負に制御することが知られている.USP8変異体はEGF受容体を過度に脱ユビキチン化しそのリソソームにおける分解を阻害した.その結果,EGFシグナルの持続的な活性化をひき起こし,これが副腎皮質刺激ホルモンの前駆体であるプロオピオメラノコルチンの遺伝子発現および副腎皮質刺激ホルモンの分泌の亢進をひき起こすと考えられた.以上のことから,下垂体の副腎皮質刺激ホルモン産生細胞におけるUSP8の機能獲得型変異がEGFシグナルの過剰をきたしCushing病をひき起こすことが示唆された.
Cushing病は脳下垂体の副腎皮質刺激ホルモン産生細胞の腺腫から副腎皮質刺激ホルモンが過剰に分泌されることによりひき起こされる1,2).副腎皮質刺激ホルモンはペプチドホルモンであり,前駆体であるプロオピオメラノコルチンが細胞において限定分解されることにより産生される.副腎皮質刺激ホルモンは副腎皮質に作用して副腎からの糖質コルチコイドの分泌を促進するため,Cushing病の患者では副腎皮質刺激ホルモンの分泌の過剰が糖質コルチコイドの分泌の過剰をひき起こす.そして,糖質コルチコイドの過剰が満月様顔貌,中心性肥満,糖尿病,高血圧などの合併症をひき起こす.Cushing病は治療しないと死にいたることもある疾患であるが,これまで有効な治療薬がなく,高度な外科的な技術を要する下垂体腺腫の切除が完治のための唯一の治療法になっている.そのため,Cushing病は厚生労働省の定める特定疾患でもあり,この難病の有効な治療薬の開発にむけその発症機構の解明が待ち望まれていた.
下垂体の副腎皮質刺激ホルモン産生細胞における副腎皮質刺激ホルモンの前駆体プロオピオメラノコルチンの遺伝子発現において,EGFからのシグナルが重要なはたらきを担うことが示唆されている3,4).EGFが結合して活性化されたEGF受容体はすみやかにエンドサイトーシスされ,エンドソームをへてリソソームへと輸送され分解される.このEGF受容体の下方制御は,活性化されたEGF受容体から発信されるシグナルが過剰になることで細胞の過剰な応答をひき起こすことを防ぐためのしくみである.このとき,活性化されたEGF受容体はユビキチン化され,それがリソソームへの輸送シグナルとしてはたらく5,6).筆者らのグループは,活性化されエンドサイトーシスされたEGF受容体をエンドソームにおいて脱ユビキチン化酵素USP8が脱ユビキチン化し,リソソーム輸送シグナルを停止することによりその下方制御を抑制することを明らかにしていた7).筆者らのグループはまた,USP8の14-3-3結合モチーフであるArg-Ser-Tyr-Ser-Ser-Proに14-3-3タンパク質が結合することにより,その脱ユビキチン化活性が抑制されることを見い出していた8).
今回,10人のCushing病の患者から摘出した下垂体腫瘍の全エキソーム解析を行った.その結果,4人の腫瘍においてUSP8の14-3-3結合モチーフに変異が見い出された.これらの変異USP8遺伝子は野生型のUSP8遺伝子と混在し,また,同じ患者のほか組織からは検出されなかったことから,ヘテロ体細胞変異であると考えられた.さらに,別の7人のCushing病の患者の下垂体腫瘍においてUSP8遺伝子の塩基配列を調べた結果,2人の腫瘍において同様の変異がみつかり,計17人中6人(35%)の腫瘍においてUSP8の14-3-3結合モチーフにホットスポット変異がみつかった(図1).

USP8の14-3-3結合モチーフの変異がCushing病をひき起こす分子機構について調べるため,見い出されたUSP8変異体を生化学的および細胞生物学的に解析した.おのおののUSP8変異体において14-3-3との結合能を調べたところ,4つのUSP8変異体では結合がほぼ完全に消失し,1つのUSP8変異体ではまだ少し結合能が残っていた.FLAGタグを付加してCOS-7細胞に発現させ抗FLAG抗体により精製したおのおののUSP8変異体を用いて,EGF受容体に対する脱ユビキチン化活性を調べた結果,少し結合能が残っていたUSP8変異体のほかは,野生型USP8より脱ユビキチン化活性が上昇していることがわかった.しかし,その上昇のレベルはUSP8変異体ごとにまちまちであり,野生型より顕著に高い活性を示さないUSP8変異体も存在した.
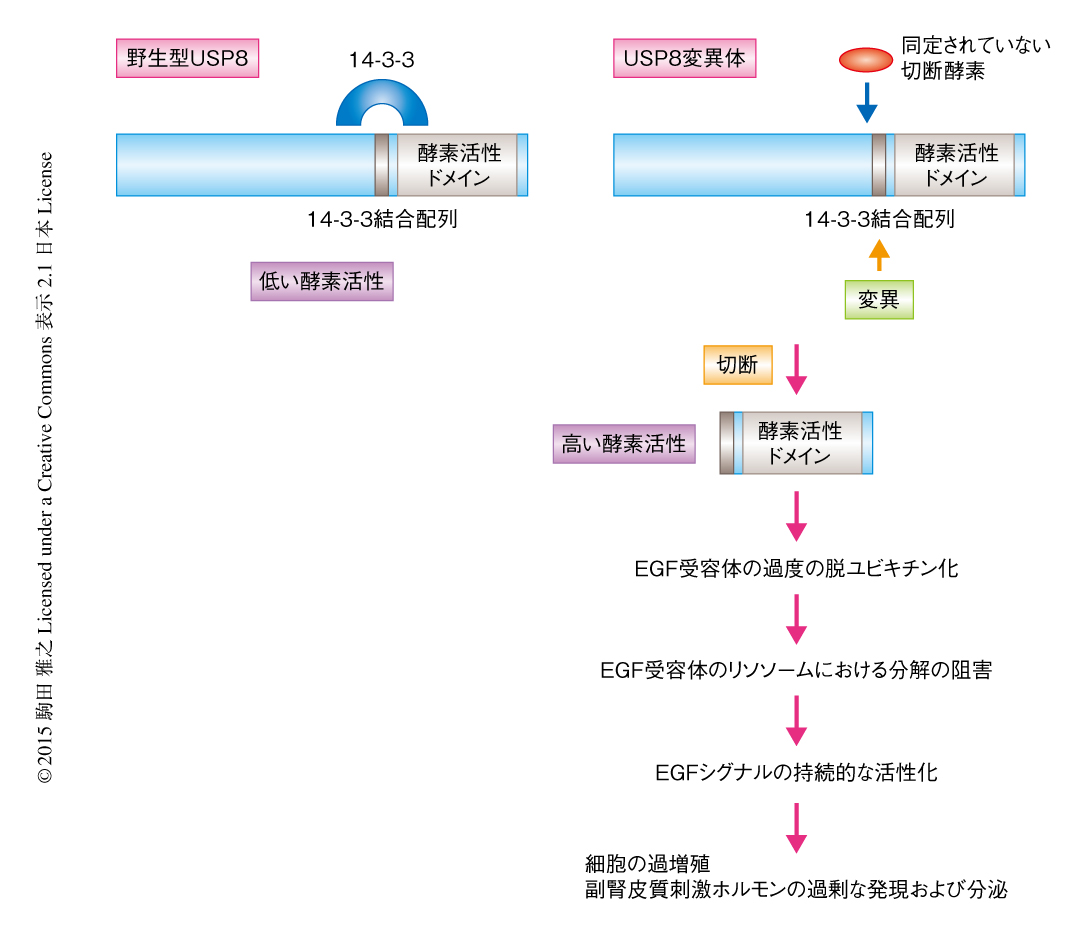
このとき,USP8変異体には130 kDaの全長型にくわえ,マイナーな90 kDaの短縮型の検出されることに気づいた.この短縮型は野生型USP8の場合にはほとんど検出されず,興味深いことに,短縮型の量の多い変異体ほどEGF受容体に対する脱ユビキチン化活性が高くなっていた.野生型USP8と比べ脱ユビキチン化活性の上昇のみられなかった1つのUSP変異体では,短縮型はまったく検出されなかった.USP8変異体を精製し電気泳動ののちクマシーブリリアントブルー染色をしたところ,90 kDaの短縮型にくわえ40 kDaのバンドが検出された.したがって,USP8変異体では130 kDaの全長タンパク質が90 kDaと40 kDaの断片に限定分解されやすくなっていることがわかった.これらのバンドをゲルから切り出しトリプシンにより消化して質量分析を行った結果,この限定分解は14-3-3結合モチーフの直前で起こっていることがわかった.その結果として生じるC末端側の40 kDaの断片(C40)は,ほぼ酵素活性ドメインのみからなっていた.FLAGタグを付加したC40をCOS-7細胞から免疫精製し,その脱ユビキチン化活性を全長の野生型USP8と比較した結果,C40は過剰に高い脱ユビキチン化活性を獲得していることが明らかになった.すなわち,14-3-3結合モチーフに変異をもつUSP8は14-3-3と結合できなくなった結果,その結合配列の近傍で限定分解されやすくなり,限定分解により生じた酵素活性ドメインの断片であるC40が過剰な脱ユビキチン化活性をもつことがわかった(図2).
USP8変異体あるいは限定分解により生じたC40を発現させたCOS-7細胞をEGFにより刺激し,EGF受容体のユビキチン化について調べた結果,USP8変異体およびC40は細胞においても活性化EGF受容体を野生型USP8より強く脱ユビキチン化することがわかった.ヒトのHeLa細胞の免疫蛍光染色によりEGFの刺激から1時間のちにおけるEGF受容体の細胞内局在を調べたところ,野生型USP8を発現させた細胞では正常な細胞と同様に,EGF受容体はエンドソームあるいはリソソームに取り込まれていた.これに対し,USP8変異体あるいはC40を発現させた細胞ではEGF受容体の細胞膜への顕著な蓄積が観察された.低分子量GTPaseであるRab11のGDP結合型変異体を発現させてエンドサイトーシスされたEGF受容体の細胞膜へのリサイクリングを阻害したところ,C40を発現させた細胞ではEGF受容体がリサイクリングエンドソームに蓄積した.これらの結果から,USP8変異体およびC40はいちどエンドサイトーシスされた活性化EGF受容体をエンドソームにおいて過度に脱ユビキチン化することにより,細胞膜へのリサイクリングを亢進することが示唆された.
EGFの刺激はMAPキナーゼであるERKをリン酸化して活性化することにより下流にシグナルを伝達する.USP8変異体およびC40を発現させた細胞をEGFにより2時間の刺激をしたときのERKのリン酸化をリン酸化ERKに対する抗体による免疫蛍光染色により調べたところ,正常な細胞および野生型USP8を過剰に発現させた細胞ではERKのリン酸化のレベルは非常に低かったのに対し,USP8変異体あるいはC40を発現させた細胞ではERKのリン酸化が残存しており,USP8遺伝子の変異が活性化EGF受容体からの持続的なEGFシグナルの発信をひき起こすことが示唆された.
USP8遺伝子の変異が副腎皮質刺激ホルモン産生細胞における副腎皮質刺激ホルモンの産生および分泌に影響するかどうかを,マウスの下垂体の副腎皮質刺激ホルモン産生細胞株であるAtT-20株を用いて調べた.その結果,USP8変異体あるいはC40の発現は,EGFシグナルに依存した副腎皮質刺激ホルモンの前駆体プロオピオメラノコルチンの遺伝子のプロモーター活性,および,副腎皮質刺激ホルモンの細胞外への分泌を亢進した.プロオピオメラノコルチンの遺伝子のプロモーター活性の上昇は,EGFの刺激により活性化される転写因子AP-1を介することが示唆された.一方,少なくとも今回の実験系では,USP8変異体あるいはC40がEGFの刺激によるAtT-20株の増殖を顕著に亢進するようすはみられなかった.したがって,USP8遺伝子の変異がひき起こす下垂体の副腎皮質刺激ホルモン産生細胞におけるEGFシグナルの持続的な活性化は,細胞の増殖より副腎皮質刺激ホルモンの過剰な分泌への寄与が大きいのではないかと考えられた.
この研究により,USP8の14-3-3結合モチーフの変異が過剰活性化変異となりCushing病の引き金になること,そして,その活性化はUSP8の限定分解によることが示された(図2).これらの結果は,USP8およびまだ同定されていないUSP8の切断酵素が,これまで存在しなかったCushing病の有効な治療薬の開発にむけた新たな分子標的となりうることを提示するものであった.
しかし,限定分解による活性化をうけないUSP8変異体も1つみつかっており,そのようなUSP8遺伝子の変異による発症機構の解明は残された課題である.さらに,USP8遺伝子の変異がみつかったのはCushing病の患者全体の約1/3であり,残り2/3の患者における発症機構の解明はより大きな課題である.
略歴:1994年 東京大学大学院薬学系研究科にて博士号取得,関西医科大学 助手,1996年 米国Fred Hutchinson Cancer Research Center博士研究員,2000年 東京工業大学大学院生命理工学研究科 助手,2001年 同 助教授を経て,2014年より同 教授.
研究テーマ:ユビキチン化による細胞膜タンパク質のリソソームへの輸送および分解の制御機構.
抱負:ユビキチンによる細胞機能制御の基礎研究もつづけたいが,今後は,Cushing病の治療にむけた医学的な応用研究もはじめたい.
研究室URL:http://www.komada-lab.bio.titech.ac.jp/
© 2015 駒田 雅之 Licensed under CC 表示 2.1 日本
(東京工業大学大学院生命理工学研究科 生体システム専攻情報生物学分野)
email:駒田雅之
DOI: 10.7875/first.author.2015.002
Mutations in the deubiquitinase gene USP8 cause Cushing's disease.
Martin Reincke, Silviu Sbiera, Akira Hayakawa, Marily Theodoropoulou, Andrea Osswald, Felix Beuschlein, Thomas Meitinger, Emi Mizuno-Yamasaki, Kohei Kawaguchi, Yasushi Saeki, Keiji Tanaka, Thomas Wieland, Elisabeth Graf, Wolfgang Saeger, Cristina L. Ronchi, Bruno Allolio, Michael Buchfelder, Tim M. Strom, Martin Fassnacht, Masayuki Komada
Nature Genetics, 47, 31-38 (2015)
要 約
Cushing病は脳下垂体の副腎皮質刺激ホルモン産生細胞の腺腫からの副腎皮質刺激ホルモンの過剰な分泌が原因となり発症する.10人のCushing病の患者から摘出した下垂体腫瘍の全エキソーム解析の結果,4人の腫瘍において脱ユビキチン化酵素USP8の遺伝子に体細胞変異が見い出された.これらの変異はUSP8の14-3-3結合モチーフに集中しており,変異を生じたUSP8は14-3-3タンパク質との結合能を失っていた.その結果,USP8変異体は14-3-3結合モチーフの直前で限定分解をうけやすくなり,限定分解により生じたC末端側の酵素活性ドメインのみからなる断片は高い脱ユビキチン化活性を示した.USP8はEGFにより活性化されたEGF受容体を脱ユビキチン化し,そのリソソームへの輸送および分解を負に制御することが知られている.USP8変異体はEGF受容体を過度に脱ユビキチン化しそのリソソームにおける分解を阻害した.その結果,EGFシグナルの持続的な活性化をひき起こし,これが副腎皮質刺激ホルモンの前駆体であるプロオピオメラノコルチンの遺伝子発現および副腎皮質刺激ホルモンの分泌の亢進をひき起こすと考えられた.以上のことから,下垂体の副腎皮質刺激ホルモン産生細胞におけるUSP8の機能獲得型変異がEGFシグナルの過剰をきたしCushing病をひき起こすことが示唆された.
はじめに
Cushing病は脳下垂体の副腎皮質刺激ホルモン産生細胞の腺腫から副腎皮質刺激ホルモンが過剰に分泌されることによりひき起こされる1,2).副腎皮質刺激ホルモンはペプチドホルモンであり,前駆体であるプロオピオメラノコルチンが細胞において限定分解されることにより産生される.副腎皮質刺激ホルモンは副腎皮質に作用して副腎からの糖質コルチコイドの分泌を促進するため,Cushing病の患者では副腎皮質刺激ホルモンの分泌の過剰が糖質コルチコイドの分泌の過剰をひき起こす.そして,糖質コルチコイドの過剰が満月様顔貌,中心性肥満,糖尿病,高血圧などの合併症をひき起こす.Cushing病は治療しないと死にいたることもある疾患であるが,これまで有効な治療薬がなく,高度な外科的な技術を要する下垂体腺腫の切除が完治のための唯一の治療法になっている.そのため,Cushing病は厚生労働省の定める特定疾患でもあり,この難病の有効な治療薬の開発にむけその発症機構の解明が待ち望まれていた.
1.Cushing病におけるUSP8の14-3-3結合モチーフのホットスポット変異
下垂体の副腎皮質刺激ホルモン産生細胞における副腎皮質刺激ホルモンの前駆体プロオピオメラノコルチンの遺伝子発現において,EGFからのシグナルが重要なはたらきを担うことが示唆されている3,4).EGFが結合して活性化されたEGF受容体はすみやかにエンドサイトーシスされ,エンドソームをへてリソソームへと輸送され分解される.このEGF受容体の下方制御は,活性化されたEGF受容体から発信されるシグナルが過剰になることで細胞の過剰な応答をひき起こすことを防ぐためのしくみである.このとき,活性化されたEGF受容体はユビキチン化され,それがリソソームへの輸送シグナルとしてはたらく5,6).筆者らのグループは,活性化されエンドサイトーシスされたEGF受容体をエンドソームにおいて脱ユビキチン化酵素USP8が脱ユビキチン化し,リソソーム輸送シグナルを停止することによりその下方制御を抑制することを明らかにしていた7).筆者らのグループはまた,USP8の14-3-3結合モチーフであるArg-Ser-Tyr-Ser-Ser-Proに14-3-3タンパク質が結合することにより,その脱ユビキチン化活性が抑制されることを見い出していた8).
今回,10人のCushing病の患者から摘出した下垂体腫瘍の全エキソーム解析を行った.その結果,4人の腫瘍においてUSP8の14-3-3結合モチーフに変異が見い出された.これらの変異USP8遺伝子は野生型のUSP8遺伝子と混在し,また,同じ患者のほか組織からは検出されなかったことから,ヘテロ体細胞変異であると考えられた.さらに,別の7人のCushing病の患者の下垂体腫瘍においてUSP8遺伝子の塩基配列を調べた結果,2人の腫瘍において同様の変異がみつかり,計17人中6人(35%)の腫瘍においてUSP8の14-3-3結合モチーフにホットスポット変異がみつかった(図1).
2.限定分解によるUSP8変異体の活性化
USP8の14-3-3結合モチーフの変異がCushing病をひき起こす分子機構について調べるため,見い出されたUSP8変異体を生化学的および細胞生物学的に解析した.おのおののUSP8変異体において14-3-3との結合能を調べたところ,4つのUSP8変異体では結合がほぼ完全に消失し,1つのUSP8変異体ではまだ少し結合能が残っていた.FLAGタグを付加してCOS-7細胞に発現させ抗FLAG抗体により精製したおのおののUSP8変異体を用いて,EGF受容体に対する脱ユビキチン化活性を調べた結果,少し結合能が残っていたUSP8変異体のほかは,野生型USP8より脱ユビキチン化活性が上昇していることがわかった.しかし,その上昇のレベルはUSP8変異体ごとにまちまちであり,野生型より顕著に高い活性を示さないUSP8変異体も存在した.
このとき,USP8変異体には130 kDaの全長型にくわえ,マイナーな90 kDaの短縮型の検出されることに気づいた.この短縮型は野生型USP8の場合にはほとんど検出されず,興味深いことに,短縮型の量の多い変異体ほどEGF受容体に対する脱ユビキチン化活性が高くなっていた.野生型USP8と比べ脱ユビキチン化活性の上昇のみられなかった1つのUSP変異体では,短縮型はまったく検出されなかった.USP8変異体を精製し電気泳動ののちクマシーブリリアントブルー染色をしたところ,90 kDaの短縮型にくわえ40 kDaのバンドが検出された.したがって,USP8変異体では130 kDaの全長タンパク質が90 kDaと40 kDaの断片に限定分解されやすくなっていることがわかった.これらのバンドをゲルから切り出しトリプシンにより消化して質量分析を行った結果,この限定分解は14-3-3結合モチーフの直前で起こっていることがわかった.その結果として生じるC末端側の40 kDaの断片(C40)は,ほぼ酵素活性ドメインのみからなっていた.FLAGタグを付加したC40をCOS-7細胞から免疫精製し,その脱ユビキチン化活性を全長の野生型USP8と比較した結果,C40は過剰に高い脱ユビキチン化活性を獲得していることが明らかになった.すなわち,14-3-3結合モチーフに変異をもつUSP8は14-3-3と結合できなくなった結果,その結合配列の近傍で限定分解されやすくなり,限定分解により生じた酵素活性ドメインの断片であるC40が過剰な脱ユビキチン化活性をもつことがわかった(図2).
3.USP8変異体によるEGF受容体の下方制御の阻害
USP8変異体あるいは限定分解により生じたC40を発現させたCOS-7細胞をEGFにより刺激し,EGF受容体のユビキチン化について調べた結果,USP8変異体およびC40は細胞においても活性化EGF受容体を野生型USP8より強く脱ユビキチン化することがわかった.ヒトのHeLa細胞の免疫蛍光染色によりEGFの刺激から1時間のちにおけるEGF受容体の細胞内局在を調べたところ,野生型USP8を発現させた細胞では正常な細胞と同様に,EGF受容体はエンドソームあるいはリソソームに取り込まれていた.これに対し,USP8変異体あるいはC40を発現させた細胞ではEGF受容体の細胞膜への顕著な蓄積が観察された.低分子量GTPaseであるRab11のGDP結合型変異体を発現させてエンドサイトーシスされたEGF受容体の細胞膜へのリサイクリングを阻害したところ,C40を発現させた細胞ではEGF受容体がリサイクリングエンドソームに蓄積した.これらの結果から,USP8変異体およびC40はいちどエンドサイトーシスされた活性化EGF受容体をエンドソームにおいて過度に脱ユビキチン化することにより,細胞膜へのリサイクリングを亢進することが示唆された.
EGFの刺激はMAPキナーゼであるERKをリン酸化して活性化することにより下流にシグナルを伝達する.USP8変異体およびC40を発現させた細胞をEGFにより2時間の刺激をしたときのERKのリン酸化をリン酸化ERKに対する抗体による免疫蛍光染色により調べたところ,正常な細胞および野生型USP8を過剰に発現させた細胞ではERKのリン酸化のレベルは非常に低かったのに対し,USP8変異体あるいはC40を発現させた細胞ではERKのリン酸化が残存しており,USP8遺伝子の変異が活性化EGF受容体からの持続的なEGFシグナルの発信をひき起こすことが示唆された.
4.USP8変異体による副腎皮質刺激ホルモンの産生および分泌の亢進
USP8遺伝子の変異が副腎皮質刺激ホルモン産生細胞における副腎皮質刺激ホルモンの産生および分泌に影響するかどうかを,マウスの下垂体の副腎皮質刺激ホルモン産生細胞株であるAtT-20株を用いて調べた.その結果,USP8変異体あるいはC40の発現は,EGFシグナルに依存した副腎皮質刺激ホルモンの前駆体プロオピオメラノコルチンの遺伝子のプロモーター活性,および,副腎皮質刺激ホルモンの細胞外への分泌を亢進した.プロオピオメラノコルチンの遺伝子のプロモーター活性の上昇は,EGFの刺激により活性化される転写因子AP-1を介することが示唆された.一方,少なくとも今回の実験系では,USP8変異体あるいはC40がEGFの刺激によるAtT-20株の増殖を顕著に亢進するようすはみられなかった.したがって,USP8遺伝子の変異がひき起こす下垂体の副腎皮質刺激ホルモン産生細胞におけるEGFシグナルの持続的な活性化は,細胞の増殖より副腎皮質刺激ホルモンの過剰な分泌への寄与が大きいのではないかと考えられた.
おわりに
この研究により,USP8の14-3-3結合モチーフの変異が過剰活性化変異となりCushing病の引き金になること,そして,その活性化はUSP8の限定分解によることが示された(図2).これらの結果は,USP8およびまだ同定されていないUSP8の切断酵素が,これまで存在しなかったCushing病の有効な治療薬の開発にむけた新たな分子標的となりうることを提示するものであった.
しかし,限定分解による活性化をうけないUSP8変異体も1つみつかっており,そのようなUSP8遺伝子の変異による発症機構の解明は残された課題である.さらに,USP8遺伝子の変異がみつかったのはCushing病の患者全体の約1/3であり,残り2/3の患者における発症機構の解明はより大きな課題である.
文 献
- Cushing, H.: The basophil adenomas of the pituitary body and their clinical manifestations. Bull. Johns Hopkins Hosp., 50, 127-195 (1932)
- Melmed, S.: Pathogenesis of pituitary tumors. Nat. Rev. Endocrinol., 7, 257-266 (2011)[PubMed]
- Theodoropoulou, M., Arzberger, T., Gruebler, Y. et al.: Expression of epidermal growth factor receptor in neoplastic pituitary cells: evidence for a role in corticotropinoma cells. J. Endocrinol., 183, 385-394 (2004)[PubMed]
- Fukuoka, H., Cooper, O., Ben-Shlomo, A. et al.: EGFR as a therapeutic target for human, canine, and mouse ACTH-secreting pituitary adenomas. J. Clin. Invest., 121, 4712-4721 (2011)[PubMed]
- Clague, M. J., Liu, H. & Urbe, S.: Governance of endocytic trafficking and signaling by reversible ubiquitylation. Dev. Cell, 23, 457-467 (2012)[PubMed]
- Tanno, H. & Komada, M.: The ubiquitin code and its decoding machinery in the endocytic pathway. J. Biochem., 153, 497-504 (2013)[PubMed]
- Mizuno, E., Iura, T., Mukai, A. et al.: Regulation of epidermal growth factor receptor down-regulation by UBPY-mediated deubiquitination at endosomes. Mol. Biol. Cell, 16, 5163-5174 (2005)[PubMed]
- Mizuno, E., Kitamura, N. & Komada, M.: 14-3-3-dependent inhibition of the deubiquitinating activity of UBPY and its cancellation in the M phase. Exp. Cell Res., 313, 3624-3634 (2007)[PubMed]
著者プロフィール
略歴:1994年 東京大学大学院薬学系研究科にて博士号取得,関西医科大学 助手,1996年 米国Fred Hutchinson Cancer Research Center博士研究員,2000年 東京工業大学大学院生命理工学研究科 助手,2001年 同 助教授を経て,2014年より同 教授.
研究テーマ:ユビキチン化による細胞膜タンパク質のリソソームへの輸送および分解の制御機構.
抱負:ユビキチンによる細胞機能制御の基礎研究もつづけたいが,今後は,Cushing病の治療にむけた医学的な応用研究もはじめたい.
研究室URL:http://www.komada-lab.bio.titech.ac.jp/
© 2015 駒田 雅之 Licensed under CC 表示 2.1 日本
