DNA損傷に依存するクロマチンのユビキチン化のOTUB1による通常とは異なる抑制
中田 慎一郎
(慶應義塾大学医学部 総合医科研究センター咸臨丸プロジェクト)
email:中田慎一郎
DOI: 10.7875/first.author.2010.024
Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1.
Shinichiro Nakada, Ikue Tai, Stephanie Panier, Abdallah Al-Hakim, Shun-ichiro Iemura, Yu-Chi Juang, Lara O’Donnell, Ayako Kumakubo, Meagan Munro, Frank Sicheri, Anne-Claude Gingras, Tohru Natsume, Toshio Suda, Daniel Durocher
Nature, 466, 941-946 (2010)
DNA損傷のなかでもっとも危険なDNA 2本鎖損傷では,ユビキチンリガーゼであるRNF8やRNF168などによるクロマチンのユビキチン化を介したシグナル伝達系により,DNA修復や細胞周期チェックポイントの制御が行われている.ここでは,脱ユビキチン化酵素OTUB1によるクロマチンのユビキチン化の抑制機構を報告する.OTUB1は,RNF168とともにはたらくユビキチン結合酵素UBC13と結合し,ユビキチン鎖の切断ではなく,ユビキチンのイソペプチド結合をさまたげることによってユビキチン鎖の形成を抑制している.また,DNA損傷応答の最上流タンパク質であるATMが阻害された際に減弱するDNA損傷応答がOTUB1の発現抑制により回復することから,OTUB1とUBC13との結合はDNA損傷応答を増強するための薬剤ターゲットとなる可能性が示唆される.
ユビキチン化は重要なタンパク質の翻訳後修飾である.ユビキチン化反応は,ATP依存的に行われるユビキチン活性化酵素(E1)へのユビキチンのチオエステル結合,ユビキチン結合酵素(E2)へのユビキチンの転移(このときも,チオエステル結合),そして,ユビキチンリガーゼ(E3)を介した基質へのユビキチンの転移(このときは,イソペプチド結合),というカスケードによって行われる.多くのユビキチンリガーゼ自体にはユビキチンの転移は行われない.ユビキチンには7つのリジン残基が含まれており,このうちのひとつにほかのユビキチンのC末端がイソペプチド結合することをくり返すことによってユビキチン鎖が形成される.最近,ユビキチンのどのリジン残基が使われるかによりユビキチン鎖の役割が異なっていることがわかってきた.
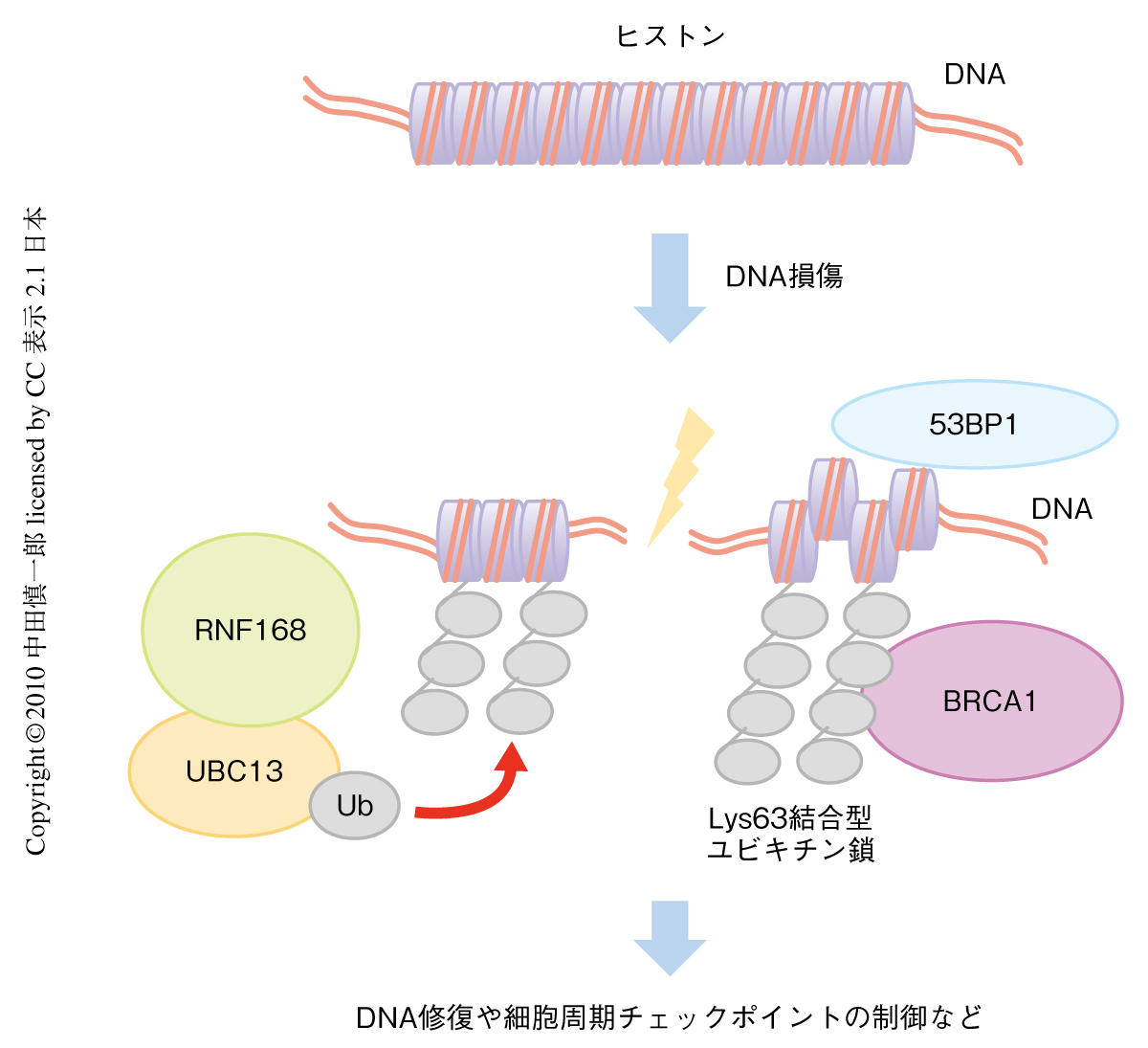
DNA損傷応答にかかわる遺伝子変異は,放射線高感受性の高発がん性遺伝性疾患,たとえば,家族性乳がん(責任遺伝子は,BRCA1遺伝子とBRCA2遺伝子)や毛細血管拡張性運動失調症(責任遺伝子は,ATM遺伝子)の原因となることが知られている.DNA 2本鎖損傷部位ではクロマチン結合タンパク質のリン酸化とユビキチン化が起こり,DNA修復や細胞周期チェックポイント制御のためのシグナル伝達が行われる.リン酸化はおもにセリン-スレオニンキナーゼであるATMタンパク質によって行われており,ユビキチン化はユビキチン結合酵素UBC13と,ユビキチンリガーゼRNF8 1-3) およびRNF168 4,5) により行われている.DNA損傷部位の周囲で形成されるユビキチン鎖は,プロテアソーム系での分解の目印となるLys48結合型ユビキチン鎖とは異なるLys63結合型ユビキチン鎖であり,このユビキチン鎖には,RAP80,Abraxas,BRCA1からなるタンパク質複合体が結合する.また,Lys63結合型ユビキチン鎖が付加される基質としてヒストンH2Aが知られている.ヒストンのユビキチン化にともなってクロマチンの立体構造の変化が起こると考えられており,これにより,メチル化ヒストンH4への53BP1の結合が促進される.DNA損傷部位へのBRCA1や53BP1のリクルートがうまくいかないと,DNA修復や細胞周期チェックポイントに異常が生じる(図1).
DNA損傷のないとき(あるいは,DNA修復ののち)にはクロマチンのユビキチン化は抑制されなければならないが,その分子機構についてはほとんど解明されていなかった.そこで筆者らは,このユビキチン化の抑制機構の解明のための研究を行った.
DNA損傷に依存したクロマチンのユビキチン化の抑制タンパク質を発見するため,まず,ユビキチン化抑制タンパク質としてもっとも可能性の高い脱ユビキチン化酵素を対象としたsiRNAスクリーニングを行った.脱ユビキチン化酵素の触媒ドメインは,その構造から,USPドメイン,UCHドメイン,OTUドメイン,MJDドメイン,JAMMドメインの5つに分類される6).まず,このなかのOTUファミリーを対象とした.siRNAを遺伝子導入した骨肉腫由来細胞株U2OSに3 Gyの放射線を照射し,細胞内でのユビキチン鎖形成の経時変化を結合型ユビキチンに対する抗体FK2を用いた蛍光免疫染色法によりモニターした.
このスクリーニングにより,OTUB1の発現を抑制した細胞ではDNA損傷依存的に形成されるユビキチン鎖が長時間にわたって持続することが見い出された.さらに,OTUB1特異的なsiRNAを遺伝子導入した細胞ではクロマチンのユビキチン化に依存して生じる53BP1の電離放射線誘発フォーカス(ionizing radiation induced foci)も持続的に観察された.一方,遺伝子導入によりOTUB1を一過的に過剰発現させた場合には,ヒストンH2AXのリン酸化や,MDC1,RNF8,RNF168のDNA損傷部位への再局在は影響されず,結合型ユビキチンの形成および53BP1の電離放射線誘発フォーカスの形成が阻害された.これらの結果より,OTUB1はRNF168のかかわるクロマチンのユビキチン化を抑制していることが示された.
最近,OTUB1は脱ユビキチン化酵素としてLys48結合型ユビキチン鎖に強い特異性をもつことが報告され7,8),OTUB1がDNA損傷応答において形成されるLys63結合型ユビキチン鎖の切断を行っていることに疑念が生じた.そこで,OTUドメインの脱活性中心Cys91をSerに置換した変異体OTUB1(C91S)の過剰発現がDNA損傷応答を阻害できるのかどうかを調べた.すると,驚いたことに,OTUB1(C91S)変異体はクロマチンのユビキチン化も53BP1の電離放射線誘発フォーカスの形成も阻害した.一方,OTUサブファミリーのなかでOTUB1と構造が非常に似ているOTUB2や,OTUB2とは相同性のないN末端の46アミノ酸残基を欠いた変異体OTUB1ΔN,OTUドメインの触媒3つ組残基Asp88,Cys91,His265をそれぞれAla,Ser,Alaと置換した変異体OTUB1(ASA)の過剰発現では,クロマチンのユビキチン化は阻害されなかった.
この免疫染色から得た結果がユビキチンリガーゼRNF168に依存したヒストンのユビキチン化を反映したものであるかどうかを確認するため,293T細胞にRNF168とユビキチンを過剰発現させてヒストンをユビキチン化するin vitroユビキチン化アッセイを行った.RNF168およびユビキチンとともに,野生型OTUB1,OTUB1(C91S)変異体,OTUB1ΔN変異体,あるいは,OTUB1(ASA)変異体を過剰発現させると,やはり,野生型OTUB1とOTUB1(C91S)変異体だけがヒストンのユビキチン化を阻害した.これらの結果を総合すると,DNA損傷依存性ユビキチン化はOTUB1により抑制されるが,これは,クロマチンにおけるユビキチン鎖の切断とは異なる機構によるものと考えられた.
OTUB1が直接に結合してその機能を制御しているタンパク質を同定するため,免疫沈降-液体クロマトグラフィー-タンデム質量分析計(IP-LC-MS/MS)を用いてOTUB1結合タンパク質を検索した.その解析結果から,OTUB1は,UBE2Dファミリー,UBE2Eファミリー,UBC13(UBE2N)といったユビキチン結合酵素や,ユビキチン自体と複合体を形成していることが判明した.そこで,RNF168とともにクロマチンのユビキチン化を担っていることが知られているユビキチン結合酵素UBC13に焦点をあてて解析を進めた.
OTUB1およびその変異体とUBC13との結合を免疫沈降法で解析したところ,クロマチンのユビキチン化を阻害できる野生型OTUB1とOTUB1(C91S)変異体はUBC13と結合し,クロマチンのユビキチン化を阻害できないOTUB1ΔN変異体とOTUB1(ASA)変異体はUBC13とは結合しないことが判明した.さらに,組換えタンパク質のみを用いたプルダウンアッセイによりOTUB1とUBC13とは直接に結合することが示された.だだし,プルダウンアッセイの結果は免疫沈降の結果とはやや異なっていて,免疫沈降ではUBC13との結合が検出されなかったOTUB1ΔN変異体も,弱いながらUBC13と結合することが示された.OTUB1(ASA)変異体はプルダウンアッセイにおいてもUBC13とは結合しなかった.
最近の報告で,OTUB1のN末端にはOTUドメインとは異なる第2のユビキチン結合部位があることが示されていたため8),OTUB1のN末端にはUBC13に結合したユビキチンと結合する(ユビキチン化反応の中間体として,ユビキチンはUBC13のCys87にチオエステル結合する)のではないかと考え,ユビキチンの結合したUBC13をもちいてOTUB1によるプルダウンアッセイを行った.結果は予想を肯定するもので,野生型OTUB1はユビキチンが結合していないUBC13よりもユビキチンが結合したUBC13と強く結合し,OTUB1ΔN変異体はユビキチンが結合したUBC13とは結合しなかった.
つぎに,組換えタンパク質のみを用いてRNF168とUBC13とによるユビキチン鎖の形成反応をin vitroで再現し,OTUB1とUBC13との結合が直接,ユビキチン鎖の形成を阻害するのかどうかを確認した.このアッセイでは,ユビキチン活性化酵素,UBC13,および,UBC13とヘテロ複合体を形成するユビキチン結合酵素バリアントUEV1aのみでも短いユビキチン鎖が形成され,さらに,RNF168を添加することによりユビキチン鎖の伸長反応が促進されて長いユビキチン鎖が形成された.野生型OTUB1はRNF168存在下であっても非存在下であってもユビキチン鎖の形成を完全に阻害した.これに対し,UBC13と結合しないOTUB1(ASA)変異体はユビキチン鎖の形成を阻害しなかった.
これらの結果を総合すると,OTUB1はユビキチンの結合したUBC13と結合し,ユビキチン鎖の形成を阻害していると結論できた.
では,OTUB1はどのようにしてユビキチン鎖の形成を抑制しているのだろうか.理論上,3つの可能性,つまりOTUB1が,1)UBC13とユビキチンとのチオエステル結合の形成(ユビキチンの結合)を阻害する,2)UBC13にチオエステル結合したユビキチンの解離(ユビキチンの解離)を阻害する.3)UBC13から解離したユビキチンがほかのユビキチンにイソペプチド結合するのを阻害する,があるので,それぞれの可能性について検証実験を行った.
まず,ユビキチン活性化酵素UBE1,UBC13とユビキチンをATP存在化でインキュベーションし,UBC13にユビキチンを結合させるという実験を行った.還元剤なしの条件でSDS-ポリアクリルアミドゲル電気泳動を行い抗UBC13抗体によりウェスタンブロットを行うと,UBC13のほかに,UBC13よりもユビキチン1分子分だけ泳動距離が短い位置にバンドが観察された.このバンドは還元剤処理により消失することから,UBC13にユビキチンがチオエステル結合したものであることが示された.組換えOTUB1をくわえてこのアッセイを行ってもユビキチンの結合したUBC13が検出された.つまり,OTUB1はUBC13へのユビキチン結合を阻害していないことが示された.この結果は,ユビキチンの結合したUBC13とOTUB1とが結合するというデータとあいいれるものだった.
つぎに,同様の方法でUBC13にビオチン化ユビキチンを結合し,ユビキチン結合酵素バリアントUEV1aと過剰量のビオチン化されていないユビキチンを添加した.OTUB1ΔN変異体が存在する条件でこの反応を進めるとUBC13からビオチン化ユビキチンの解離が進み,一方で,ユビキチン2分子結合体の生成が進んだ.これに対し,野生型OTUB1あるいはOTUB1(C91S)変異体が存在するときには,UBC13からのビオチン化ユビキチンの解離は進むものの,ユビキチン2分子結合体の形成は認められなかった.OTUB1ΔN変異体は脱ユビキチン活性を保っており,OTUB1(C91S)変異体は脱ユビキチン活性を失っていることから,OTUB1のユビキチン2分子結合体の形成阻害はユビキチン鎖の切断とは異なる機構によるものと結論づけられた.
さらに,OTUB1が前述のIP-LC-MS/MSにより検出されたUBE2D/UBE2Eサブファミリーのユビキチン活性化酵素の機能も抑制するかどうかを調べた.免疫沈降法により,OTUB1は細胞内でUBE2D2と結合することが示された.さらに,組換えタンパク質を用いたプルダウンアッセイにより,UBC13の場合と同様に,OTUB1はユビキチンの結合したUBE2D2により強い親和性をもつことが示された.in vitroユビキチン化アッセイでは,脱ユビキチン活性をもたないOTUB1(C91S)変異体がUBE2D2とユビキチンリガーゼTRAF6とによるユビキチン鎖の形成を抑制し,さらに,UBE2D3とTRAF6,UBE2D2とユビキチンリガーゼIpaHによるユビキチン鎖の形成を抑制することが示された.一方,IP-LC-MS/MSでは検出されなかったユビキチン結合酵素UBE2L3とユビキチンリガーゼSMURFによるユビキチン鎖の形成はOTUB1により抑制されなかった.これらの結果から,OTUB1は少なくともin vitroにおいて,UBC13に対するのと同じ機構でUBE2D/2Eサブファミリーユビキチン結合酵素の機能を抑制することがわかった.
ATMはDNA損傷応答の最上流ではたらくキナーゼであり,これを薬剤により阻害するとDNA損傷応答シグナル伝達系が減弱し,53BP1の電離放射線誘発フォーカスが小さくなる9).また,DNA修復のひとつである相同組換えの効率が低下する10).ATM阻害を行う細胞であらかじめOTUB1のノックダウンを行っておくと,53BP1の電離放射線誘発フォーカスの大きさが回復し,さらに,相同組換えの効率も回復した.逆に,OTUB1を過剰発現させた細胞では相同組換えの効率が低下した.これは,OTUB1の過剰発現によりBRCA1の電離放射線誘発フォーカスの形成が阻害されたことによるものと考えられた.
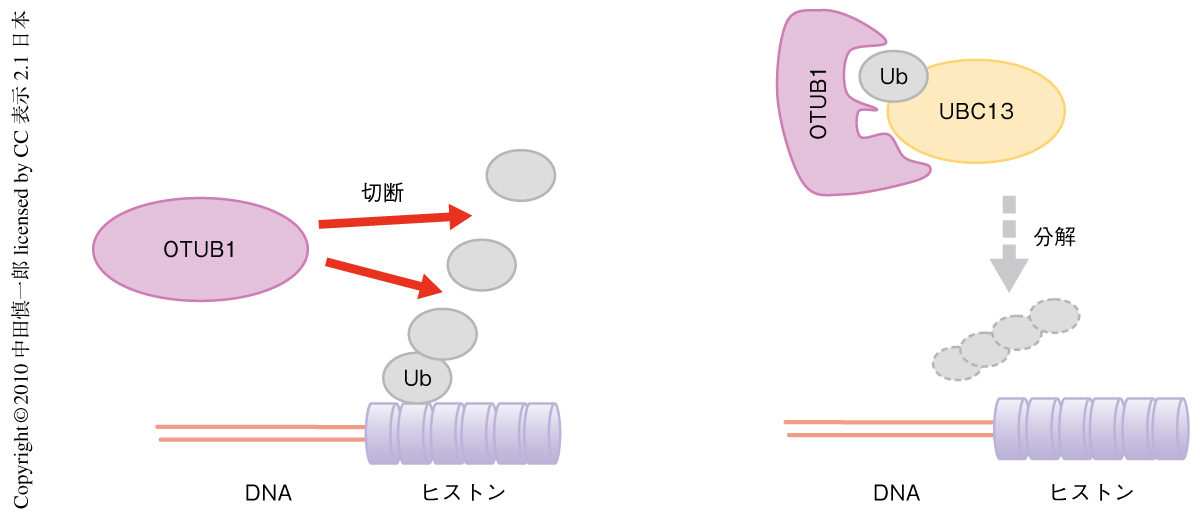
OTUB1によるDNA損傷依存性のクロマチンのユビキチン化の抑制は,ユビキチン鎖の分解ではなく,UBC13依存性のユビキチン鎖形成の直接阻害であった(図2).この阻害機構にはOTUB1の脱ユビキチン活性は必要ない.OTUB1とUBC13との結合にはOTUドメインとN末端に存在の予想されているユビキチン結合ドメインが必要であると考えられる.電離放射線誘発フォーカス減少のようすやOTUB1の機能から,OTUB1はDNA損傷応答におけるユビキチン化の閾値を設定しているものと考えられる.
脱ユビキチン活性に非依存的に発揮されるOTUB1の機能は,脱ユビキチン化酵素の新しい機能を示唆するものである.脱ユビキチン化酵素のなかには脱ユビキチン活性を発揮する構造をもっていないもののあることが知られており11),これらの脱ユビキチン化酵素にも新たな機能が備わっているのかもしれない.
また,OTUB1とUBC13の結合を阻害することで,たとえば,毛細血管拡張性運動失調症のようにDNA損傷応答が減弱している疾患のDNA損傷応答を回復させることが期待でき,OTUB1とUBC13の相互作用は将来的な薬剤ターゲットとなりうる.
略歴:2005年 東京医科歯科大学大学大学院医歯学総合研究科 修了,2006年 カナダMount Sinai Hospital Post Doctoral Fellowを経て,2009年より慶應義塾大学医学部 特別研究講師.
研究テーマ:DNA損傷応答,染色体不安定性.
関心事:発がん予防法開発への分子生物学的な根拠の提示.
© 2010 中田 慎一郎 Licensed under CC 表示 2.1 日本
(慶應義塾大学医学部 総合医科研究センター咸臨丸プロジェクト)
email:中田慎一郎
DOI: 10.7875/first.author.2010.024
Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1.
Shinichiro Nakada, Ikue Tai, Stephanie Panier, Abdallah Al-Hakim, Shun-ichiro Iemura, Yu-Chi Juang, Lara O’Donnell, Ayako Kumakubo, Meagan Munro, Frank Sicheri, Anne-Claude Gingras, Tohru Natsume, Toshio Suda, Daniel Durocher
Nature, 466, 941-946 (2010)
要 約
DNA損傷のなかでもっとも危険なDNA 2本鎖損傷では,ユビキチンリガーゼであるRNF8やRNF168などによるクロマチンのユビキチン化を介したシグナル伝達系により,DNA修復や細胞周期チェックポイントの制御が行われている.ここでは,脱ユビキチン化酵素OTUB1によるクロマチンのユビキチン化の抑制機構を報告する.OTUB1は,RNF168とともにはたらくユビキチン結合酵素UBC13と結合し,ユビキチン鎖の切断ではなく,ユビキチンのイソペプチド結合をさまたげることによってユビキチン鎖の形成を抑制している.また,DNA損傷応答の最上流タンパク質であるATMが阻害された際に減弱するDNA損傷応答がOTUB1の発現抑制により回復することから,OTUB1とUBC13との結合はDNA損傷応答を増強するための薬剤ターゲットとなる可能性が示唆される.
はじめに
ユビキチン化は重要なタンパク質の翻訳後修飾である.ユビキチン化反応は,ATP依存的に行われるユビキチン活性化酵素(E1)へのユビキチンのチオエステル結合,ユビキチン結合酵素(E2)へのユビキチンの転移(このときも,チオエステル結合),そして,ユビキチンリガーゼ(E3)を介した基質へのユビキチンの転移(このときは,イソペプチド結合),というカスケードによって行われる.多くのユビキチンリガーゼ自体にはユビキチンの転移は行われない.ユビキチンには7つのリジン残基が含まれており,このうちのひとつにほかのユビキチンのC末端がイソペプチド結合することをくり返すことによってユビキチン鎖が形成される.最近,ユビキチンのどのリジン残基が使われるかによりユビキチン鎖の役割が異なっていることがわかってきた.
DNA損傷応答にかかわる遺伝子変異は,放射線高感受性の高発がん性遺伝性疾患,たとえば,家族性乳がん(責任遺伝子は,BRCA1遺伝子とBRCA2遺伝子)や毛細血管拡張性運動失調症(責任遺伝子は,ATM遺伝子)の原因となることが知られている.DNA 2本鎖損傷部位ではクロマチン結合タンパク質のリン酸化とユビキチン化が起こり,DNA修復や細胞周期チェックポイント制御のためのシグナル伝達が行われる.リン酸化はおもにセリン-スレオニンキナーゼであるATMタンパク質によって行われており,ユビキチン化はユビキチン結合酵素UBC13と,ユビキチンリガーゼRNF8 1-3) およびRNF168 4,5) により行われている.DNA損傷部位の周囲で形成されるユビキチン鎖は,プロテアソーム系での分解の目印となるLys48結合型ユビキチン鎖とは異なるLys63結合型ユビキチン鎖であり,このユビキチン鎖には,RAP80,Abraxas,BRCA1からなるタンパク質複合体が結合する.また,Lys63結合型ユビキチン鎖が付加される基質としてヒストンH2Aが知られている.ヒストンのユビキチン化にともなってクロマチンの立体構造の変化が起こると考えられており,これにより,メチル化ヒストンH4への53BP1の結合が促進される.DNA損傷部位へのBRCA1や53BP1のリクルートがうまくいかないと,DNA修復や細胞周期チェックポイントに異常が生じる(図1).
DNA損傷のないとき(あるいは,DNA修復ののち)にはクロマチンのユビキチン化は抑制されなければならないが,その分子機構についてはほとんど解明されていなかった.そこで筆者らは,このユビキチン化の抑制機構の解明のための研究を行った.
1.OTUB1はRNF168依存性のDNA損傷応答を制御している
DNA損傷に依存したクロマチンのユビキチン化の抑制タンパク質を発見するため,まず,ユビキチン化抑制タンパク質としてもっとも可能性の高い脱ユビキチン化酵素を対象としたsiRNAスクリーニングを行った.脱ユビキチン化酵素の触媒ドメインは,その構造から,USPドメイン,UCHドメイン,OTUドメイン,MJDドメイン,JAMMドメインの5つに分類される6).まず,このなかのOTUファミリーを対象とした.siRNAを遺伝子導入した骨肉腫由来細胞株U2OSに3 Gyの放射線を照射し,細胞内でのユビキチン鎖形成の経時変化を結合型ユビキチンに対する抗体FK2を用いた蛍光免疫染色法によりモニターした.
このスクリーニングにより,OTUB1の発現を抑制した細胞ではDNA損傷依存的に形成されるユビキチン鎖が長時間にわたって持続することが見い出された.さらに,OTUB1特異的なsiRNAを遺伝子導入した細胞ではクロマチンのユビキチン化に依存して生じる53BP1の電離放射線誘発フォーカス(ionizing radiation induced foci)も持続的に観察された.一方,遺伝子導入によりOTUB1を一過的に過剰発現させた場合には,ヒストンH2AXのリン酸化や,MDC1,RNF8,RNF168のDNA損傷部位への再局在は影響されず,結合型ユビキチンの形成および53BP1の電離放射線誘発フォーカスの形成が阻害された.これらの結果より,OTUB1はRNF168のかかわるクロマチンのユビキチン化を抑制していることが示された.
2.OTUB1はUBC13と結合して脱ユビキチン化活性に非依存的にクロマチンのユビキチン化を抑制する
最近,OTUB1は脱ユビキチン化酵素としてLys48結合型ユビキチン鎖に強い特異性をもつことが報告され7,8),OTUB1がDNA損傷応答において形成されるLys63結合型ユビキチン鎖の切断を行っていることに疑念が生じた.そこで,OTUドメインの脱活性中心Cys91をSerに置換した変異体OTUB1(C91S)の過剰発現がDNA損傷応答を阻害できるのかどうかを調べた.すると,驚いたことに,OTUB1(C91S)変異体はクロマチンのユビキチン化も53BP1の電離放射線誘発フォーカスの形成も阻害した.一方,OTUサブファミリーのなかでOTUB1と構造が非常に似ているOTUB2や,OTUB2とは相同性のないN末端の46アミノ酸残基を欠いた変異体OTUB1ΔN,OTUドメインの触媒3つ組残基Asp88,Cys91,His265をそれぞれAla,Ser,Alaと置換した変異体OTUB1(ASA)の過剰発現では,クロマチンのユビキチン化は阻害されなかった.
この免疫染色から得た結果がユビキチンリガーゼRNF168に依存したヒストンのユビキチン化を反映したものであるかどうかを確認するため,293T細胞にRNF168とユビキチンを過剰発現させてヒストンをユビキチン化するin vitroユビキチン化アッセイを行った.RNF168およびユビキチンとともに,野生型OTUB1,OTUB1(C91S)変異体,OTUB1ΔN変異体,あるいは,OTUB1(ASA)変異体を過剰発現させると,やはり,野生型OTUB1とOTUB1(C91S)変異体だけがヒストンのユビキチン化を阻害した.これらの結果を総合すると,DNA損傷依存性ユビキチン化はOTUB1により抑制されるが,これは,クロマチンにおけるユビキチン鎖の切断とは異なる機構によるものと考えられた.
OTUB1が直接に結合してその機能を制御しているタンパク質を同定するため,免疫沈降-液体クロマトグラフィー-タンデム質量分析計(IP-LC-MS/MS)を用いてOTUB1結合タンパク質を検索した.その解析結果から,OTUB1は,UBE2Dファミリー,UBE2Eファミリー,UBC13(UBE2N)といったユビキチン結合酵素や,ユビキチン自体と複合体を形成していることが判明した.そこで,RNF168とともにクロマチンのユビキチン化を担っていることが知られているユビキチン結合酵素UBC13に焦点をあてて解析を進めた.
OTUB1およびその変異体とUBC13との結合を免疫沈降法で解析したところ,クロマチンのユビキチン化を阻害できる野生型OTUB1とOTUB1(C91S)変異体はUBC13と結合し,クロマチンのユビキチン化を阻害できないOTUB1ΔN変異体とOTUB1(ASA)変異体はUBC13とは結合しないことが判明した.さらに,組換えタンパク質のみを用いたプルダウンアッセイによりOTUB1とUBC13とは直接に結合することが示された.だだし,プルダウンアッセイの結果は免疫沈降の結果とはやや異なっていて,免疫沈降ではUBC13との結合が検出されなかったOTUB1ΔN変異体も,弱いながらUBC13と結合することが示された.OTUB1(ASA)変異体はプルダウンアッセイにおいてもUBC13とは結合しなかった.
最近の報告で,OTUB1のN末端にはOTUドメインとは異なる第2のユビキチン結合部位があることが示されていたため8),OTUB1のN末端にはUBC13に結合したユビキチンと結合する(ユビキチン化反応の中間体として,ユビキチンはUBC13のCys87にチオエステル結合する)のではないかと考え,ユビキチンの結合したUBC13をもちいてOTUB1によるプルダウンアッセイを行った.結果は予想を肯定するもので,野生型OTUB1はユビキチンが結合していないUBC13よりもユビキチンが結合したUBC13と強く結合し,OTUB1ΔN変異体はユビキチンが結合したUBC13とは結合しなかった.
つぎに,組換えタンパク質のみを用いてRNF168とUBC13とによるユビキチン鎖の形成反応をin vitroで再現し,OTUB1とUBC13との結合が直接,ユビキチン鎖の形成を阻害するのかどうかを確認した.このアッセイでは,ユビキチン活性化酵素,UBC13,および,UBC13とヘテロ複合体を形成するユビキチン結合酵素バリアントUEV1aのみでも短いユビキチン鎖が形成され,さらに,RNF168を添加することによりユビキチン鎖の伸長反応が促進されて長いユビキチン鎖が形成された.野生型OTUB1はRNF168存在下であっても非存在下であってもユビキチン鎖の形成を完全に阻害した.これに対し,UBC13と結合しないOTUB1(ASA)変異体はユビキチン鎖の形成を阻害しなかった.
これらの結果を総合すると,OTUB1はユビキチンの結合したUBC13と結合し,ユビキチン鎖の形成を阻害していると結論できた.
3.OTUB1はユビキチン鎖の形成におけるイソペプチド結合を阻害する
では,OTUB1はどのようにしてユビキチン鎖の形成を抑制しているのだろうか.理論上,3つの可能性,つまりOTUB1が,1)UBC13とユビキチンとのチオエステル結合の形成(ユビキチンの結合)を阻害する,2)UBC13にチオエステル結合したユビキチンの解離(ユビキチンの解離)を阻害する.3)UBC13から解離したユビキチンがほかのユビキチンにイソペプチド結合するのを阻害する,があるので,それぞれの可能性について検証実験を行った.
まず,ユビキチン活性化酵素UBE1,UBC13とユビキチンをATP存在化でインキュベーションし,UBC13にユビキチンを結合させるという実験を行った.還元剤なしの条件でSDS-ポリアクリルアミドゲル電気泳動を行い抗UBC13抗体によりウェスタンブロットを行うと,UBC13のほかに,UBC13よりもユビキチン1分子分だけ泳動距離が短い位置にバンドが観察された.このバンドは還元剤処理により消失することから,UBC13にユビキチンがチオエステル結合したものであることが示された.組換えOTUB1をくわえてこのアッセイを行ってもユビキチンの結合したUBC13が検出された.つまり,OTUB1はUBC13へのユビキチン結合を阻害していないことが示された.この結果は,ユビキチンの結合したUBC13とOTUB1とが結合するというデータとあいいれるものだった.
つぎに,同様の方法でUBC13にビオチン化ユビキチンを結合し,ユビキチン結合酵素バリアントUEV1aと過剰量のビオチン化されていないユビキチンを添加した.OTUB1ΔN変異体が存在する条件でこの反応を進めるとUBC13からビオチン化ユビキチンの解離が進み,一方で,ユビキチン2分子結合体の生成が進んだ.これに対し,野生型OTUB1あるいはOTUB1(C91S)変異体が存在するときには,UBC13からのビオチン化ユビキチンの解離は進むものの,ユビキチン2分子結合体の形成は認められなかった.OTUB1ΔN変異体は脱ユビキチン活性を保っており,OTUB1(C91S)変異体は脱ユビキチン活性を失っていることから,OTUB1のユビキチン2分子結合体の形成阻害はユビキチン鎖の切断とは異なる機構によるものと結論づけられた.
4.OTUB1はUBE2D/UBE2EサブファミリーのOTUB1と結合しその機能を抑制する
さらに,OTUB1が前述のIP-LC-MS/MSにより検出されたUBE2D/UBE2Eサブファミリーのユビキチン活性化酵素の機能も抑制するかどうかを調べた.免疫沈降法により,OTUB1は細胞内でUBE2D2と結合することが示された.さらに,組換えタンパク質を用いたプルダウンアッセイにより,UBC13の場合と同様に,OTUB1はユビキチンの結合したUBE2D2により強い親和性をもつことが示された.in vitroユビキチン化アッセイでは,脱ユビキチン活性をもたないOTUB1(C91S)変異体がUBE2D2とユビキチンリガーゼTRAF6とによるユビキチン鎖の形成を抑制し,さらに,UBE2D3とTRAF6,UBE2D2とユビキチンリガーゼIpaHによるユビキチン鎖の形成を抑制することが示された.一方,IP-LC-MS/MSでは検出されなかったユビキチン結合酵素UBE2L3とユビキチンリガーゼSMURFによるユビキチン鎖の形成はOTUB1により抑制されなかった.これらの結果から,OTUB1は少なくともin vitroにおいて,UBC13に対するのと同じ機構でUBE2D/2Eサブファミリーユビキチン結合酵素の機能を抑制することがわかった.
5.OTUB1のノックダウンによりATM阻害により生じる相同組換えの異常が回復する
ATMはDNA損傷応答の最上流ではたらくキナーゼであり,これを薬剤により阻害するとDNA損傷応答シグナル伝達系が減弱し,53BP1の電離放射線誘発フォーカスが小さくなる9).また,DNA修復のひとつである相同組換えの効率が低下する10).ATM阻害を行う細胞であらかじめOTUB1のノックダウンを行っておくと,53BP1の電離放射線誘発フォーカスの大きさが回復し,さらに,相同組換えの効率も回復した.逆に,OTUB1を過剰発現させた細胞では相同組換えの効率が低下した.これは,OTUB1の過剰発現によりBRCA1の電離放射線誘発フォーカスの形成が阻害されたことによるものと考えられた.
おわりに
OTUB1によるDNA損傷依存性のクロマチンのユビキチン化の抑制は,ユビキチン鎖の分解ではなく,UBC13依存性のユビキチン鎖形成の直接阻害であった(図2).この阻害機構にはOTUB1の脱ユビキチン活性は必要ない.OTUB1とUBC13との結合にはOTUドメインとN末端に存在の予想されているユビキチン結合ドメインが必要であると考えられる.電離放射線誘発フォーカス減少のようすやOTUB1の機能から,OTUB1はDNA損傷応答におけるユビキチン化の閾値を設定しているものと考えられる.
脱ユビキチン活性に非依存的に発揮されるOTUB1の機能は,脱ユビキチン化酵素の新しい機能を示唆するものである.脱ユビキチン化酵素のなかには脱ユビキチン活性を発揮する構造をもっていないもののあることが知られており11),これらの脱ユビキチン化酵素にも新たな機能が備わっているのかもしれない.
また,OTUB1とUBC13の結合を阻害することで,たとえば,毛細血管拡張性運動失調症のようにDNA損傷応答が減弱している疾患のDNA損傷応答を回復させることが期待でき,OTUB1とUBC13の相互作用は将来的な薬剤ターゲットとなりうる.
文 献
- Kolas, N. K., Chapman, J. R., Nakada, S. et al.: Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science, 318, 1637-1640 (2007)[PubMed]
- Huen, M. S., Grant, R., Manke, I. et al.: RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell, 131, 901-914 (2007)[PubMed]
- Mailand, N., Bekker-Jensen, S., Faustrup, H. et al.: RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell, 131, 887-900 (2007)[PubMed]
- Stewart, G. S., Panier, S., Townsend, K. et al.: The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell, 136, 420-434 (2009)[PubMed]
- Doil, C., Mailand, N., Bekker-Jensen, S. et al.: RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell, 136, 435-446 (2009)[PubMed]
- Nijman, S. M., Luna-Vargas, M. P., Velds, A. et al.: A genomic and functional inventory of deubiquitinating enzymes. Cell, 123, 773-786 (2005)[PubMed]
- Edelmann, M. J., Iphofer, A., Akutsu, M. et al.: Structural basis and specificity of human otubain 1-mediateddeubiquitination. Biochem. J., 418, 379-390 (2009)[PubMed]
- Wang, T., Yin, L., Cooper, E. M. et al.: Evidence for bidentate substrate binding as the basis for the K48linkage specificity of otubain 1. J. Mol. Biol., 386, 1011-1023 (2009)[PubMed]
- Galanty, Y., Belotserkovskaya, R., Coates, J. et al.: Mammalian SUMO E3-ligases PIAS1 and PIAS4 promoteresponses to DNA double-strand breaks. Nature, 462, 935-939 (2009)[PubMed]
- Beucher, A., Birraux, J., Tchouandong, L. et al.: ATM and Artemis promote homologous recombination ofradiation-induced DNA double-strand breaks in G2. EMBO J., 28, 3413-3427 (2009)[PubMed]
- Hanna, J., Hathaway, N. A., Tone, Y. et al.: Deubiquitinating enzyme Ubp6 functions noncatalytically to delayproteasomal degradation. Cell, 127, 99-111 (2006)[PubMed]
著者プロフィール
略歴:2005年 東京医科歯科大学大学大学院医歯学総合研究科 修了,2006年 カナダMount Sinai Hospital Post Doctoral Fellowを経て,2009年より慶應義塾大学医学部 特別研究講師.
研究テーマ:DNA損傷応答,染色体不安定性.
関心事:発がん予防法開発への分子生物学的な根拠の提示.
© 2010 中田 慎一郎 Licensed under CC 表示 2.1 日本
