RIPK3はネクローシスに非依存的にサイトカインの産生および組織の修復を促進する
森脇健太・Francis Ka-Ming Chan
(米国Massachusetts大学Medical School,Department of Pathology)
email:森脇健太
DOI: 10.7875/first.author.2014.136
The necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair.
Kenta Moriwaki, Sakthi Balaji, Thomas McQuade, Nidhi Malhotra, Joonsoo Kang, Francis Ka-Ming Chan
Immunity, 41, 567-578 (2014)
RIPK3はプログラム型のネクローシスを制御するタンパク質であり,RIPK3により制御されるネクローシスをとくにネクロプトーシスとよぶ.ネクローシスを起こした細胞は細胞の外に免疫誘導物質を放出するため,RIPK3はさまざまな炎症性疾患においてネクロプトーシスを誘導することにより炎症反応を促進すると考えられてきた.この研究では,RIPK3によるネクロプトーシスに非依存的な炎症反応の促進能について明らかにした.RIPK3を欠損した樹状細胞ではリポ多糖の刺激ののちのサイトカインの産生がネクロプトーシスに非依存的に減少しており,これは,NF-κBシグナル伝達経路およびカスパーゼの活性化が抑制されているためであった.この樹状細胞におけるRIPK3に依存的なサイトカインの産生は,デキストラン硫酸ナトリウムにより誘導される腸炎におけるインターロイキン22を介した組織の修復に必要であり,RIPK3ノックアウトマウスはデキストラン硫酸ナトリウムによる組織への傷害を修復することができず,炎症が長期化し,野生型のマウスに比べ強い腸炎を起こした.RIPK3はネクロプトーシスに非依存的に樹状細胞におけるサイトカインの産生を促進し,傷害をうけた組織の修復を促進することが明らかになった.
アポトーシスおよびネクローシスは形態学的に異なる細胞死を表す用語であり,クロマチンの凝集および細胞の縮小化と断片化を呈す細胞死をアポトーシス,オルガネラと細胞の膨張および細胞膜の破裂を呈す細胞死をネクローシスとよぶ.発生の段階においてアポトーシスが観察されることから,アポトーシスは不必要な細胞を排除するために遺伝的にプログラムされた細胞死として認識され,これまでの数十年間,その制御機構の解明のため精力的な研究が行われ,カスパーゼをはじめとする多くの制御タンパク質が同定されてきた.それに対し,ネクローシスは物理的なストレスなどにより誘導される偶発的な非プログラム型の細胞死と考えられてきた.しかしながら,TNFなどアポトーシスを誘導するタンパク質は特定の条件のもとではネクローシスを誘導することが観察され,近年,このネクローシスを制御するタンパク質としてRIPK1,RIPK3,MLKLが同定された.このタイプのネクローシスはとくにネクロプトーシスとよばれる1).
TNFの刺激により,ある条件のもとではカスパーゼ8が活性化されてアポトーシスが誘導される.RIPK1およびRIPK3はシステインプロテアーゼであるカスパーゼ8の基質であるため,通常ではTNFの刺激ののちカスパーゼ8により切断されて不活性化される.しかし,カスパーゼ8の活性を阻害すると,TNFの刺激ののちRIPK1およびRIPK3が活性化されてネクロプトーシスが誘導される1).実際に,カスパーゼ8ノックアウトマウスあるいはカスパーゼ8のアダプタータンパク質であるFADDのノックアウトマウスは,発生の時期に激しいネクロプトーシスを起こして胎生致死となる2-4).また,カスパーゼ8あるいはFADDの腸管に特異的なノックアウトマウスはネクロプトーシスによる激しい腸炎を起こす5,6).このように,ネクロプトーシスはカスパーゼ8の活性を阻害したときにとくに強く誘導される(図1).
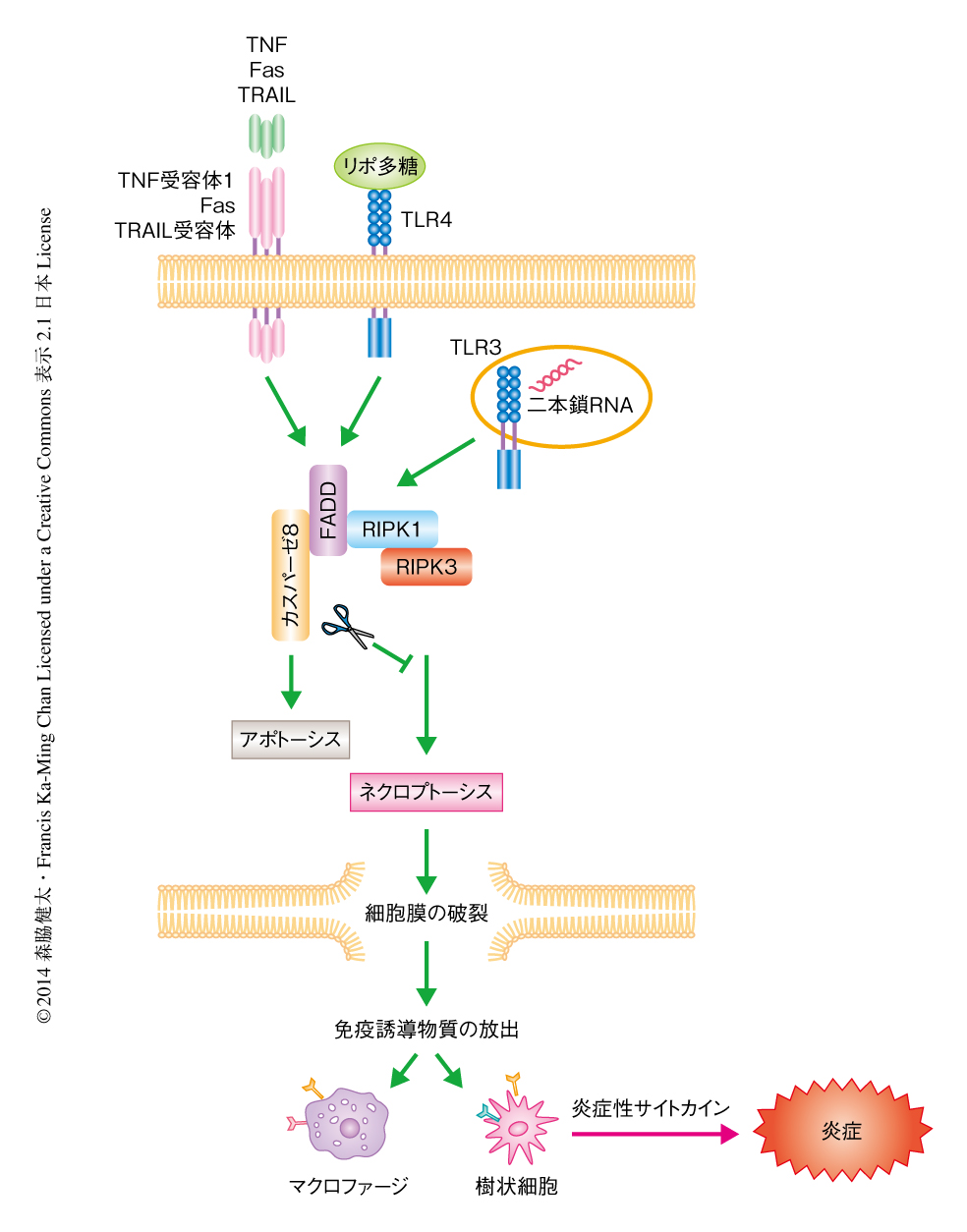
多くの炎症性疾患のモデルにおいて,RIPK3ノックアウトマウスは炎症反応が抑制されていることが報告されており,RIPK3は炎症性疾患における新たな創薬の標的として注目をあびている1).ネクローシスを起こした細胞は細胞膜が破壊されるため,免疫誘導物質を放出し周囲の免疫細胞を刺激することにより炎症反応を誘導する7)(図1).そのため,RIPK3は生体においてネクロプトーシスを起こすことにより炎症反応を誘導し,炎症性疾患に関与していると結論づけられてきた.しかしながら,in vivoにおいてネクロプトーシスを直接的に検出および定量する手法が確立されていないため,カスパーゼ8あるいはFADDを欠損していない野生型のマウスにおいて,RIPK3がネクロプトーシスを介して炎症反応を誘導しているのかどうかは明らかにされていなかった8).
潰瘍性大腸炎やクローン病に代表される炎症性腸疾患は,免疫制御機構の破綻によりひき起こされる腸管における慢性炎症のために組織が傷害される難治性の疾患である.デキストラン硫酸ナトリウムを経口投与すると腸管上皮細胞に傷害がひき起こされ腸炎が発症する.その病態が炎症性腸疾患に類似しているため,デキストラン硫酸ナトリウムにより誘導される腸炎は炎症性腸疾患のモデルとして広く利用されている.野生型のマウスおよびRIPK3ノックアウトマウスにデキストラン硫酸ナトリウムを投与して腸炎の程度を比較したところ,野生型マウスに比べRIPK3ノックアウトマウスにおいてはいちじるしい体重の減少および腸管組織の萎縮がみられ,組織学的な解析により強い炎症がみられた.デキストラン硫酸ナトリウムを投与された腸管においては,腸管上皮細胞および腸管に侵入してきた免疫細胞においてRIPK3の発現が強く誘導されていた.紫外線を照射したのちRIPK3ノックアウトマウスに由来する骨髄細胞を移植した野生型のマウスは,対照となるRIPK3ノックアウトマウスと同じ程度の腸炎を呈したことから,おもに免疫細胞に発現するRIPK3がデキストラン硫酸ナトリウムに誘導性の腸炎の抑制に重要であることがわかった.
デキストラン硫酸ナトリウムの投与ののち7日目のRIPK3ノックアウトマウスの腸管において,インターロイキン6やCox2といったいくつかの炎症性サイトカインの発現が低下していた.この時期はデキストラン硫酸ナトリウムによる組織の傷害が起こりはじめている時期であり,RIPK3ノックアウトマウスでは初期の炎症反応が抑制されていると考えられた.炎症反応は傷害をうけた組織を修復するための生体反応であり,その反応が弱すぎても強すぎても正常な組織の修復は起こらず,病態はより悪化する.デキストラン硫酸ナトリウムを投与された腸管組織においては傷害をうけた組織を修復するため腸管上皮細胞の増殖が亢進する.RIPK3ノックアウトマウスではこの腸管上皮細胞の増殖が有意に低下していたため,RIPK3は腸管における組織の修復を促進すると考えられた.
デキストラン硫酸ナトリウムに誘導性の腸炎において組織の修復を制御するサイトカインとしてインターロイキン22が知られている9).インターロイキン22はインターロイキン10ファミリーに属し,腸管が傷害されたときおもに腸管の自然リンパ球から分泌され,腸管上皮細胞に発現する受容体と結合することにより組織の修復を誘導する.RIPK3ノックアウトマウスではこのインターロイキン22の発現が有意に低下しており,その結果と一致して,インターロイキン22により誘導される転写因子STAT3のリン酸化および抗菌ペプチドReg3βの発現も低下していた.また,デキストラン硫酸ナトリウムを投与したRIPK3ノックアウトマウスに対し,インターロイキン22より生体における安定性の高いFc融合インターロイキン22を投与したところ,対照となるマウスに比べ腸炎が抑制された.自然リンパ球からのインターロイキン22の産生を誘導するサイトカインとしてインターロイキン23およびインターロイキン1βが知られているが,それらの発現はデキストラン硫酸ナトリウムを投与したRIPK3ノックアウトマウスの腸管において有意に低下していた.また,組換え体のインターロイキン23およびインターロイキン1βをRIPK3ノックアウトマウスに投与したところ,対照に比べ腸管組織におけるインターロイキン22の産生が回復し腸炎の程度が抑制された.以上の結果から,RIPK3はデキストラン硫酸ナトリウムを投与したマウスの腸管の免疫細胞においてインターロイキン23およびインターロイキン1βの産生を促進し,その結果,インターロイキン22を介した組織の修復を亢進させることにより腸炎を抑制していると考えられた(図2).
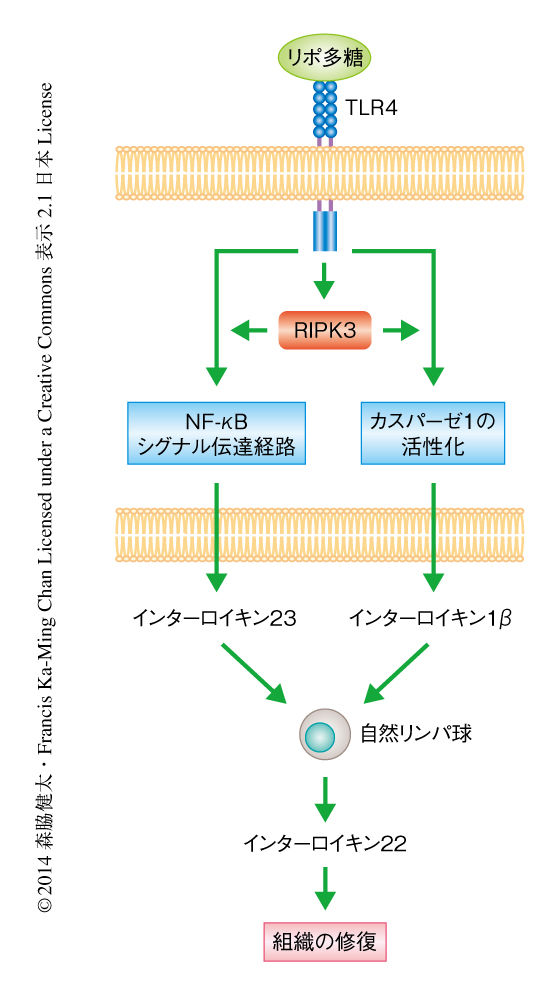
インターロイキン23およびインターロイキン1βはおもに樹状細胞,マクロファージといった自然免疫細胞において産生される.そこで,野生型のマウスに由来する骨髄細胞あるいはRIPK3ノックアウトマウスに由来する骨髄細胞を樹状細胞あるいはマクロファージへと分化させ,リポ多糖により刺激したのちのインターロイキン23の産生について調べたところ,RIPK3を欠損した骨髄に由来する樹状細胞においてインターロイキン23の産生が有意に低下していた.骨髄に由来するマクロファージにおいてはそのような差はみられなかった.骨髄に由来する樹状細胞をリポ多糖により刺激しても細胞死はみられないため,RIPK3を欠損した骨髄に由来する樹状細胞におけるインターロイキン23の産生の低下は,RIPK3の細胞死の制御機能とは関係ないものと考えられた.リポ多糖により誘導されるインターロイキン23の産生は,Toll様受容体のひとつTLR4の下流においてNF-κBシグナル伝達経路により制御されている.NF-κBシグナル伝達経路が活性化されると,NF-κBファミリータンパク質であるRelA,RelB,cRel,p50,p52が種々の組合せの二量体として核へと移行し遺伝子発現を誘導する.詳細な解析の結果,RIPK3を欠損した骨髄に由来する樹状細胞ではリポ多糖の刺激ののちのRelBおよびp50の核への移行がいちじるしく抑制されていることがわかった.それに対し,RelA,cRel,p52の核への移行は野生型の骨髄に由来する樹状細胞と同じ程度であった.RelBは非古典的NF-κBシグナル伝達経路においてp52とのヘテロ二量体としてはたらくことがよく知られているが,近年,樹状細胞ではToll様受容体により活性化される古典的NF-κBシグナル伝達経路においてp50とのヘテロ二量体としてはたらき,炎症性サイトカインの産生にかかわることが報告されている10).
野生型の骨髄に由来する樹状細胞をリポ多糖により刺激するとインターロイキン1βの産生がみられるが,このインターロイキン1βの産生もRIPK3を欠損した骨髄に由来する樹状細胞においていちじるしく抑制されていた.インターロイキン1βはインターロイキン1β前駆体として生合成され,細胞の外へと分泌されるためにはカスパーゼにより切断される必要がある.インターロイキン1β前駆体を切断するカスパーゼとしてはカスパーゼ1がもっともよく知られているが,RIPK3を欠損した骨髄に由来する樹状細胞においてはリポ多糖の刺激ののちカスパーゼ1の活性が低下していた.以上のことから,骨髄に由来する樹状細胞においてRIPK3はRelB-p50ヘテロ二量体の核への移行を促進することによりインターロイキン23の産生を誘導し,また,カスパーゼ1の活性化を促進することによりインターロイキン1βの産生を促進することがわかった(図2).
RIPK3をはじめとするネクロプトーシス制御タンパク質が同定されて以来,それらの遺伝子改変マウスを用いてネクロプトーシスと炎症性疾患との関係が明らかにされてきた.これまで,RIPK3はネクロプトーシスに特異的なタンパク質であると考えられてきたため,種々の炎症性疾患においてRIPK3はネクロプトーシスを介して炎症を誘導していると考えられてきた.しかしながら,この研究において,RIPK3がネクロプトーシスに非依存的な機能により炎症を誘導することが明らかになった.現在,RIPK3は新たな創薬の標的として注目されており,実際に,最近,RIPK3のキナーゼ活性の阻害剤が開発され,炎症性疾患への治療薬としての有用性が期待されている.RIPK3のキナーゼ活性はネクロプトーシスの誘導に必須であるため,RIPK3のキナーゼ活性の阻害剤はネクロプトーシスの阻害剤としては有用である.しかし,この研究において明らかにされたネクロプトーシスに非依存的な炎症の促進機能においてはRIPK3のキナーゼ活性は必要でないことがわかっているため,RIPK3により誘導される炎症をより効果的に抑制するためには新たな戦略が必要になるであろう.
略歴:2010年 大阪大学大学院医学系研究科 修了,同年 同 研究員を経て,2011年より米国Massachusetts大学Medical School研究員.
研究テーマ:細胞死と炎症.
Francis Ka-Ming Chan
米国Massachusetts大学Medical SchoolにてAssociate Professor.
© 2014 森脇健太・Francis Ka-Ming Chan Licensed under CC 表示 2.1 日本
(米国Massachusetts大学Medical School,Department of Pathology)
email:森脇健太
DOI: 10.7875/first.author.2014.136
The necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair.
Kenta Moriwaki, Sakthi Balaji, Thomas McQuade, Nidhi Malhotra, Joonsoo Kang, Francis Ka-Ming Chan
Immunity, 41, 567-578 (2014)
要 約
RIPK3はプログラム型のネクローシスを制御するタンパク質であり,RIPK3により制御されるネクローシスをとくにネクロプトーシスとよぶ.ネクローシスを起こした細胞は細胞の外に免疫誘導物質を放出するため,RIPK3はさまざまな炎症性疾患においてネクロプトーシスを誘導することにより炎症反応を促進すると考えられてきた.この研究では,RIPK3によるネクロプトーシスに非依存的な炎症反応の促進能について明らかにした.RIPK3を欠損した樹状細胞ではリポ多糖の刺激ののちのサイトカインの産生がネクロプトーシスに非依存的に減少しており,これは,NF-κBシグナル伝達経路およびカスパーゼの活性化が抑制されているためであった.この樹状細胞におけるRIPK3に依存的なサイトカインの産生は,デキストラン硫酸ナトリウムにより誘導される腸炎におけるインターロイキン22を介した組織の修復に必要であり,RIPK3ノックアウトマウスはデキストラン硫酸ナトリウムによる組織への傷害を修復することができず,炎症が長期化し,野生型のマウスに比べ強い腸炎を起こした.RIPK3はネクロプトーシスに非依存的に樹状細胞におけるサイトカインの産生を促進し,傷害をうけた組織の修復を促進することが明らかになった.
はじめに
アポトーシスおよびネクローシスは形態学的に異なる細胞死を表す用語であり,クロマチンの凝集および細胞の縮小化と断片化を呈す細胞死をアポトーシス,オルガネラと細胞の膨張および細胞膜の破裂を呈す細胞死をネクローシスとよぶ.発生の段階においてアポトーシスが観察されることから,アポトーシスは不必要な細胞を排除するために遺伝的にプログラムされた細胞死として認識され,これまでの数十年間,その制御機構の解明のため精力的な研究が行われ,カスパーゼをはじめとする多くの制御タンパク質が同定されてきた.それに対し,ネクローシスは物理的なストレスなどにより誘導される偶発的な非プログラム型の細胞死と考えられてきた.しかしながら,TNFなどアポトーシスを誘導するタンパク質は特定の条件のもとではネクローシスを誘導することが観察され,近年,このネクローシスを制御するタンパク質としてRIPK1,RIPK3,MLKLが同定された.このタイプのネクローシスはとくにネクロプトーシスとよばれる1).
TNFの刺激により,ある条件のもとではカスパーゼ8が活性化されてアポトーシスが誘導される.RIPK1およびRIPK3はシステインプロテアーゼであるカスパーゼ8の基質であるため,通常ではTNFの刺激ののちカスパーゼ8により切断されて不活性化される.しかし,カスパーゼ8の活性を阻害すると,TNFの刺激ののちRIPK1およびRIPK3が活性化されてネクロプトーシスが誘導される1).実際に,カスパーゼ8ノックアウトマウスあるいはカスパーゼ8のアダプタータンパク質であるFADDのノックアウトマウスは,発生の時期に激しいネクロプトーシスを起こして胎生致死となる2-4).また,カスパーゼ8あるいはFADDの腸管に特異的なノックアウトマウスはネクロプトーシスによる激しい腸炎を起こす5,6).このように,ネクロプトーシスはカスパーゼ8の活性を阻害したときにとくに強く誘導される(図1).
多くの炎症性疾患のモデルにおいて,RIPK3ノックアウトマウスは炎症反応が抑制されていることが報告されており,RIPK3は炎症性疾患における新たな創薬の標的として注目をあびている1).ネクローシスを起こした細胞は細胞膜が破壊されるため,免疫誘導物質を放出し周囲の免疫細胞を刺激することにより炎症反応を誘導する7)(図1).そのため,RIPK3は生体においてネクロプトーシスを起こすことにより炎症反応を誘導し,炎症性疾患に関与していると結論づけられてきた.しかしながら,in vivoにおいてネクロプトーシスを直接的に検出および定量する手法が確立されていないため,カスパーゼ8あるいはFADDを欠損していない野生型のマウスにおいて,RIPK3がネクロプトーシスを介して炎症反応を誘導しているのかどうかは明らかにされていなかった8).
1.RIPK3はデキストラン硫酸ナトリウムにより誘導される腸炎を抑制する
潰瘍性大腸炎やクローン病に代表される炎症性腸疾患は,免疫制御機構の破綻によりひき起こされる腸管における慢性炎症のために組織が傷害される難治性の疾患である.デキストラン硫酸ナトリウムを経口投与すると腸管上皮細胞に傷害がひき起こされ腸炎が発症する.その病態が炎症性腸疾患に類似しているため,デキストラン硫酸ナトリウムにより誘導される腸炎は炎症性腸疾患のモデルとして広く利用されている.野生型のマウスおよびRIPK3ノックアウトマウスにデキストラン硫酸ナトリウムを投与して腸炎の程度を比較したところ,野生型マウスに比べRIPK3ノックアウトマウスにおいてはいちじるしい体重の減少および腸管組織の萎縮がみられ,組織学的な解析により強い炎症がみられた.デキストラン硫酸ナトリウムを投与された腸管においては,腸管上皮細胞および腸管に侵入してきた免疫細胞においてRIPK3の発現が強く誘導されていた.紫外線を照射したのちRIPK3ノックアウトマウスに由来する骨髄細胞を移植した野生型のマウスは,対照となるRIPK3ノックアウトマウスと同じ程度の腸炎を呈したことから,おもに免疫細胞に発現するRIPK3がデキストラン硫酸ナトリウムに誘導性の腸炎の抑制に重要であることがわかった.
2.RIPK3はインターロイキン23およびインターロイキン1βの発現を介してインターロイキン22に誘導性の腸管における組織の修復を促進する
デキストラン硫酸ナトリウムの投与ののち7日目のRIPK3ノックアウトマウスの腸管において,インターロイキン6やCox2といったいくつかの炎症性サイトカインの発現が低下していた.この時期はデキストラン硫酸ナトリウムによる組織の傷害が起こりはじめている時期であり,RIPK3ノックアウトマウスでは初期の炎症反応が抑制されていると考えられた.炎症反応は傷害をうけた組織を修復するための生体反応であり,その反応が弱すぎても強すぎても正常な組織の修復は起こらず,病態はより悪化する.デキストラン硫酸ナトリウムを投与された腸管組織においては傷害をうけた組織を修復するため腸管上皮細胞の増殖が亢進する.RIPK3ノックアウトマウスではこの腸管上皮細胞の増殖が有意に低下していたため,RIPK3は腸管における組織の修復を促進すると考えられた.
デキストラン硫酸ナトリウムに誘導性の腸炎において組織の修復を制御するサイトカインとしてインターロイキン22が知られている9).インターロイキン22はインターロイキン10ファミリーに属し,腸管が傷害されたときおもに腸管の自然リンパ球から分泌され,腸管上皮細胞に発現する受容体と結合することにより組織の修復を誘導する.RIPK3ノックアウトマウスではこのインターロイキン22の発現が有意に低下しており,その結果と一致して,インターロイキン22により誘導される転写因子STAT3のリン酸化および抗菌ペプチドReg3βの発現も低下していた.また,デキストラン硫酸ナトリウムを投与したRIPK3ノックアウトマウスに対し,インターロイキン22より生体における安定性の高いFc融合インターロイキン22を投与したところ,対照となるマウスに比べ腸炎が抑制された.自然リンパ球からのインターロイキン22の産生を誘導するサイトカインとしてインターロイキン23およびインターロイキン1βが知られているが,それらの発現はデキストラン硫酸ナトリウムを投与したRIPK3ノックアウトマウスの腸管において有意に低下していた.また,組換え体のインターロイキン23およびインターロイキン1βをRIPK3ノックアウトマウスに投与したところ,対照に比べ腸管組織におけるインターロイキン22の産生が回復し腸炎の程度が抑制された.以上の結果から,RIPK3はデキストラン硫酸ナトリウムを投与したマウスの腸管の免疫細胞においてインターロイキン23およびインターロイキン1βの産生を促進し,その結果,インターロイキン22を介した組織の修復を亢進させることにより腸炎を抑制していると考えられた(図2).
3.RIPK3は骨髄に由来する樹状細胞においてリポ多糖により誘導されるNF-κBシグナル伝達経路およびカスパーゼの活性化を促進する
インターロイキン23およびインターロイキン1βはおもに樹状細胞,マクロファージといった自然免疫細胞において産生される.そこで,野生型のマウスに由来する骨髄細胞あるいはRIPK3ノックアウトマウスに由来する骨髄細胞を樹状細胞あるいはマクロファージへと分化させ,リポ多糖により刺激したのちのインターロイキン23の産生について調べたところ,RIPK3を欠損した骨髄に由来する樹状細胞においてインターロイキン23の産生が有意に低下していた.骨髄に由来するマクロファージにおいてはそのような差はみられなかった.骨髄に由来する樹状細胞をリポ多糖により刺激しても細胞死はみられないため,RIPK3を欠損した骨髄に由来する樹状細胞におけるインターロイキン23の産生の低下は,RIPK3の細胞死の制御機能とは関係ないものと考えられた.リポ多糖により誘導されるインターロイキン23の産生は,Toll様受容体のひとつTLR4の下流においてNF-κBシグナル伝達経路により制御されている.NF-κBシグナル伝達経路が活性化されると,NF-κBファミリータンパク質であるRelA,RelB,cRel,p50,p52が種々の組合せの二量体として核へと移行し遺伝子発現を誘導する.詳細な解析の結果,RIPK3を欠損した骨髄に由来する樹状細胞ではリポ多糖の刺激ののちのRelBおよびp50の核への移行がいちじるしく抑制されていることがわかった.それに対し,RelA,cRel,p52の核への移行は野生型の骨髄に由来する樹状細胞と同じ程度であった.RelBは非古典的NF-κBシグナル伝達経路においてp52とのヘテロ二量体としてはたらくことがよく知られているが,近年,樹状細胞ではToll様受容体により活性化される古典的NF-κBシグナル伝達経路においてp50とのヘテロ二量体としてはたらき,炎症性サイトカインの産生にかかわることが報告されている10).
野生型の骨髄に由来する樹状細胞をリポ多糖により刺激するとインターロイキン1βの産生がみられるが,このインターロイキン1βの産生もRIPK3を欠損した骨髄に由来する樹状細胞においていちじるしく抑制されていた.インターロイキン1βはインターロイキン1β前駆体として生合成され,細胞の外へと分泌されるためにはカスパーゼにより切断される必要がある.インターロイキン1β前駆体を切断するカスパーゼとしてはカスパーゼ1がもっともよく知られているが,RIPK3を欠損した骨髄に由来する樹状細胞においてはリポ多糖の刺激ののちカスパーゼ1の活性が低下していた.以上のことから,骨髄に由来する樹状細胞においてRIPK3はRelB-p50ヘテロ二量体の核への移行を促進することによりインターロイキン23の産生を誘導し,また,カスパーゼ1の活性化を促進することによりインターロイキン1βの産生を促進することがわかった(図2).
おわりに
RIPK3をはじめとするネクロプトーシス制御タンパク質が同定されて以来,それらの遺伝子改変マウスを用いてネクロプトーシスと炎症性疾患との関係が明らかにされてきた.これまで,RIPK3はネクロプトーシスに特異的なタンパク質であると考えられてきたため,種々の炎症性疾患においてRIPK3はネクロプトーシスを介して炎症を誘導していると考えられてきた.しかしながら,この研究において,RIPK3がネクロプトーシスに非依存的な機能により炎症を誘導することが明らかになった.現在,RIPK3は新たな創薬の標的として注目されており,実際に,最近,RIPK3のキナーゼ活性の阻害剤が開発され,炎症性疾患への治療薬としての有用性が期待されている.RIPK3のキナーゼ活性はネクロプトーシスの誘導に必須であるため,RIPK3のキナーゼ活性の阻害剤はネクロプトーシスの阻害剤としては有用である.しかし,この研究において明らかにされたネクロプトーシスに非依存的な炎症の促進機能においてはRIPK3のキナーゼ活性は必要でないことがわかっているため,RIPK3により誘導される炎症をより効果的に抑制するためには新たな戦略が必要になるであろう.
文 献
- Moriwaki, K. & Chan, F. K.: RIP3: a molecular switch for necrosis and inflammation. Genes Dev., 27, 1640-1649 (2013)[PubMed]
- Zhang, H., Zhou, X., McQuade, T. et al.: Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature, 471, 373-376 (2011)[PubMed]
- Kaiser, W. J., Upton, J. W., Long, A. B. et al.: RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature, 471, 368-372 (2011)[PubMed]
- Oberst, A., Dillon, C. P., Weinlich, R. et al.: Catalytic activity of the caspase-8-FLIPL complex inhibits RIPK3-dependent necrosis. Nature, 471, 363-367 (2011)[PubMed]
- Gunther, C., Martini, E., Wittkopf, N. et al.: Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature, 477, 335-339 (2011)[PubMed]
- Welz, P. S., Wullaert, A., Vlantis, K. et al.: FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature, 477, 330-334 (2011)[PubMed]
- Kaczmarek, A., Vandenabeele, P. & Krysko, D. V.: Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity, 38, 209-223 (2013)[PubMed]
- Moriwaki, K. & Chan, F. K.: Necrosis-dependent and independent signaling of the RIP kinases in inflammation. Cytokine Growth Factor Rev., 25, 167-174 (2014)[PubMed]
- Mizoguchi, A.: Healing of intestinal inflammation by IL-22. Inflamm. Bowel Dis., 18, 1777-1784 (2012)[PubMed]
- Shih, V. F., Davis-Turak, J., Macal, M. et al.: Control of RelB during dendritic cell activation integrates canonical and noncanonical NF-κB pathways. Nat. Immunol., 13, 1162-1170 (2012)[PubMed]
著者プロフィール
略歴:2010年 大阪大学大学院医学系研究科 修了,同年 同 研究員を経て,2011年より米国Massachusetts大学Medical School研究員.
研究テーマ:細胞死と炎症.
Francis Ka-Ming Chan
米国Massachusetts大学Medical SchoolにてAssociate Professor.
© 2014 森脇健太・Francis Ka-Ming Chan Licensed under CC 表示 2.1 日本
