アダプタータンパク質TRAF5はインターロイキン6シグナルを阻害することにより炎症性CD4陽性T細胞の分化を抑制する
長島宏行・宗 孝紀
(東北大学大学院医学系研究科 免疫学分野)
email:宗 孝紀
DOI: 10.7875/first.author.2014.046
The adaptor TRAF5 limits the differentiation of inflammatory CD4+ T cells by antagonizing signaling via the receptor for IL-6.
Hiroyuki Nagashima, Yuko Okuyama, Atsuko Asao, Takeshi Kawabe, Satoshi Yamaki, Hiroyasu Nakano, Michael Croft, Naoto Ishii, Takanori So
Nature Immunology, 15, 449-456 (2014)
アダプタータンパク質TRAF5はTNF受容体スーパーファミリーによる炎症性シグナル応答にかかわることが知られていたが,CD4陽性T細胞におけるその詳細な機能は不明であった.今回,筆者らは,TRAF5がCD4陽性T細胞の分化の過程においてインターロイキン6シグナルを抑制し,これがヘルパーT細胞のサブセットであるTh17細胞の分化に対し抑制的にはたらくという予想外のシグナル伝達機構を見い出した.すなわち,TRAF5がシグナル伝達タンパク質であるgp130と恒常的に結合することにより,インターロイキン6の刺激に依存して起こるSTAT3のgp130へのリクルートおよび活性化を阻害することを発見した.さらに,Th17細胞に依存的な自己免疫疾患モデルである実験的自己免疫性脳脊髄炎においては,TRAF5の欠損により症状が増悪することがわかった.以上より,TRAF5がインターロイキン6の刺激による炎症性ヘルパーT細胞の分化を抑制することで,自己免疫疾患の発症および病態を制限する機構が明らかにされた.
CD4陽性ヘルパーT細胞はB細胞による抗体の産生を誘導し,マクロファージやCD8陽性T細胞を活性化する.その体液性免疫および細胞性免疫に対する活性化の作用は諸刃の剣であり,病原体やがん細胞の排除にかかわる一方で,アレルギーや自己免疫疾患の病態の形成と関連する.ヘルパーT細胞の分化および機能における制御機構の解明は免疫学における重要な課題であり,現在,ヘルパーT細胞を標的にした疾患の制御に関する応用研究も精力的に行われている.
外来性の抗原とまだ接触したことのないナイーブCD4陽性T細胞は3つの異質なシグナル,すなわち,T細胞受容体シグナル,補助刺激受容体シグナル,サイトカイン受容体シグナルを受け取ることによりヘルパーT細胞へと分化する(図1).とりわけ重要なことは,受け取るサイトカイン受容体シグナルの質的な差により機能の異なるヘルパーT細胞のサブセットへと個別に分化することである(図1).Th17細胞はインターロイキン17を産生することにより好中球の感染部位への動員を促進する作用をもつ一方で,多発性硬化症,リウマチ性関節炎,乾癬,強直性脊椎炎など自己免疫性疾患の病態との関連が示唆される炎症性ヘルパーT細胞のサブセットである.このTh17細胞の分化には炎症性サイトカインであるインターロイキン6によるサイトカイン受容体シグナルが必要であるが,その詳細な分化制御の機構にはいまだ不明な点が多い.

TRAF5(tumor necrosis factor receptor-associated factor 5)はTRAF1からTRAF6までの6つのファミリータンパク質のひとつで,N末端側からC末端側へRINGドメイン,ジンクフィンガードメイン,TRAFドメインから構成される.RINGドメインおよびTRAFドメインにはそれぞれユビキチンリガーゼ活性およびアダプタータンパク質としての活性が認められ,これらによりNF-κBなどによる炎症性シグナル応答が促進される.TRAF5ノックアウトマウスは発生や生育に関しては正常であり,リンパ器官の形成やリンパ球の成熟において顕著な異常はとくに認められない1).
筆者らは,この研究にいたるまえ,TRAF5ノックアウトマウスにおいて肺組織における炎症の病態が亢進し,これがTRAF5欠損CD4陽性T細胞の過剰な反応性に起因するという予想に反した結果を得ていた2).この研究では,CD4陽性T細胞におけるTRAF5の機能を解析し,TRAF5がインターロイキン6受容体からのシグナルに対し抑制作用をもち,これにより炎症性Th17細胞の分化を制限する機構がはじめて明らかにされた.
CD4陽性T細胞におけるTRAF5の機能を調べるため,野生型マウスおよびTRAF5ノックアウトマウスからナイーブCD4陽性T細胞を精製し,抗CD3抗体および抗CD28抗体,および,各種のサイトカインの刺激により,Th1細胞サブセット,Th2細胞サブセット,Th17細胞サブセット,制御性T細胞サブセットへの分化をin vitroにおいて誘導した(図1).その結果,TRAF5欠損ナイーブCD4陽性T細胞からヘルパーT細胞への分化の過程において培養上清に高濃度のインターロイキン17およびインターロイキン21が検出され,最終的にTh17細胞への分化の亢進が認められた.この結果と一致して,Th17細胞に関連する遺伝子であるRorc遺伝子,Il17a遺伝子,Il17f遺伝子,Il23r遺伝子の発現がTRAF5欠損CD4陽性T細胞において有意に上昇することがわかった.さらに,ヘルパーT細胞への分化がより生理的に誘導されるナイーブCD4陽性T細胞と抗原提示細胞との共培養実験系においても,TRAF5欠損CD4陽性T細胞においてTh17細胞への分化の亢進が認められた.細胞増殖能,インターロイキン2の産生能,ほかのヘルパーT細胞サブセットへの分化に関して有意な差が認められなかったことから,TRAF5はTh17細胞への分化を特異的に抑制することが示唆された.
in vitroにおいて観察されたTh17細胞への分化における差が生体においても同様に観察されるかどうか調べるため,卵白アルブミン抗原に特異的なT細胞受容体を発現するCD4陽性T細胞を用いて養子移入実験を行った.野生型マウスおよびTRAF5ノックアウトマウスからナイーブCD4陽性T細胞を精製し,野生型マウスに移入し卵白アルブミンおよび完全フロイントアジュバントによりマウスを免疫したのち,1週間後に所属リンパ節におけるTh17細胞への分化について評価した.その結果,ドナーとなった細胞にしめるインターロイキン17産生細胞の割合および絶対数がTRAF5欠損CD4陽性細胞において有意に増加していたことから,生体におけるヘルパーT細胞への分化においてもTRAF5がTh17細胞への分化に対し抑制的にはたらくことが明らかになった.
Th17細胞はインターロイキン6およびTGFβの刺激により,制御性T細胞はTGFβの刺激により分化する(図1).制御性T細胞への分化あるいはTGFβシグナルに対するTRAF5の欠損の影響はほとんど認められなかったことから,TRAF5にインターロイキン6シグナルに対する制御能のあることが示唆された.インターロイキン6はインターロイキン6受容体(CD126)とgp130(CD130)から構成される受容体に結合することによりgp130の二量体化を促進し,gp130の細胞内領域に結合するキナーゼであるJAKを活性化し,gp130の細胞内領域のリン酸化を誘導する.STAT3はリン酸化したgp130と結合することによりリン酸化され,二量体化したのち核へと移行し転写因子としての機能を発揮する(図2).インターロイキン6シグナル伝達経路に対するTRAF5の機能を調べる目的で,野生型マウスおよびTRAF5ノックアウトマウスからCD4陽性T細胞を精製し,インターロイキン6の刺激により誘導されるSTAT3のリン酸化のレベルを比較した.その結果,TRAF5の欠損によりSTAT3のリン酸化が有意に亢進することが判明した.また,CD8陽性T細胞においても同様の結果が得られた.B細胞やマクロファージなどTRAF5あるいはgp130,または,その両方の発現レベルの低い細胞では,TRAF5のSTAT3リン酸化に対する抑制能はほとんど検出できなかった.また,インターロイキン6と同様にSTAT3のリン酸化を誘導するインターロイキン10やインターロイキン21によりCD4陽性T細胞を刺激しても,TRAF5の欠損による影響は認められなかった.これら一連の実験から,TRAF5がインターロイキン6受容体を介したSTAT3のリン酸化を特異的に抑制し,しかも,その効果は細胞種によらず,TRAF5とgp130の両者の発現量に依存することがわかった.
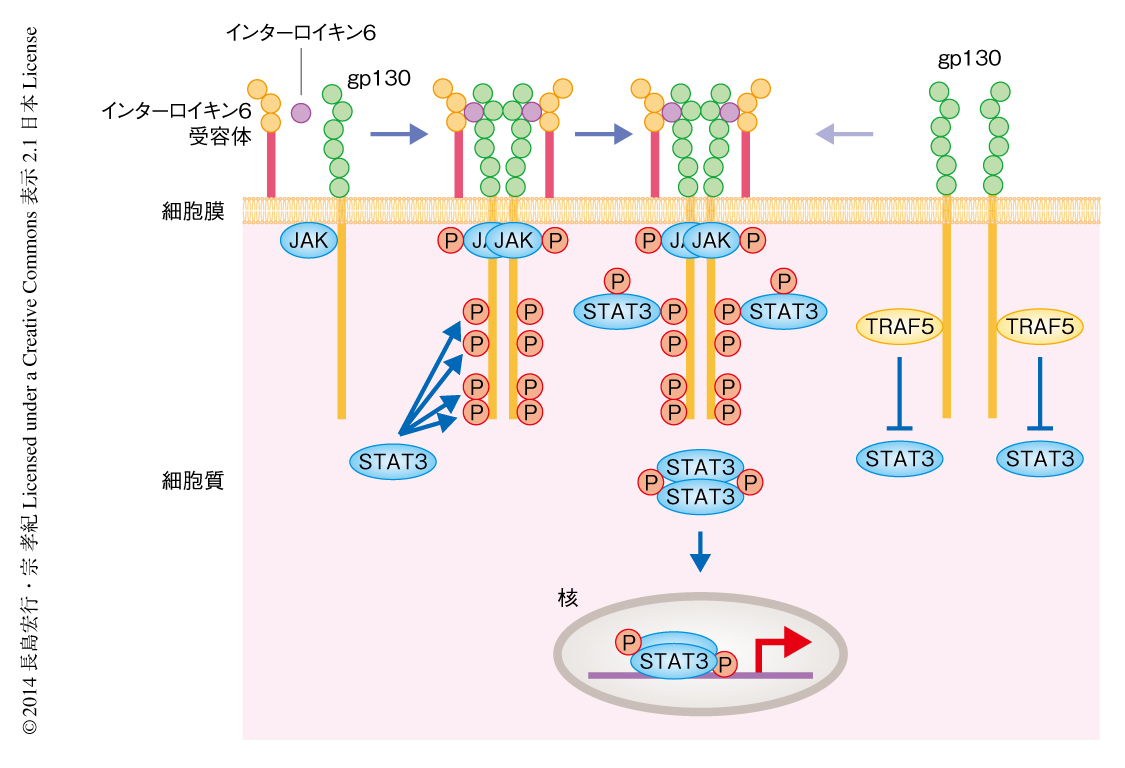
TRAF5によるインターロイキン6シグナルの抑制機構を調べる目的で,培養細胞にgp130,STAT3,TRAF5を一過性に発現させて,この3つのタンパク質の相互作用を免疫沈降法により評価した.その結果,TRAF5がインターロイキン6の刺激によらず恒常的にgp130と結合することが判明した.また,TRAF5の存在下ではインターロイキン6の刺激に依存的に起こるSTAT3とgp130とのあいだの相互作用が阻害された.重要なことに,TRAF5とgp130とのあいだの恒常的な結合はマウスから精製したCD4陽性T細胞においても検出され,この結合が生理的な環境においても起こることがわかった.これらの実験から,TRAF5がgp130に結合することによりSTAT3のgp130へのリクルートを阻害し,その結果,STAT3の活性化が抑制されるという機構が明らかになった(図2).
TRAF5とgp130がそれぞれどの領域を介して結合するかを調べた.RINGドメインおよびジンクフィンガードメインを含むTRAF5欠失変異体,および,TRAFドメインを含むTRAF5欠失変異体を作製し,gp130との結合能について評価した.その結果,TRAFドメインを含むTRAF5欠失変異体にgp130との結合活性が認められた.gp130についても細胞内領域のさまざまな欠失変異体や部位特異的な変異体を作製しTRAF5との相互作用を調べた.その結果,TRAF5がgp130の細胞内領域のアミノ酸配列774~798に存在する2つの領域と結合することが判明した.この領域の近傍にはSTAT3との結合部位が存在したことから,TRAF5とgp130とのあいだの結合がSTAT3とgp130とのあいだの結合に対し立体障害的にはたらくことにより,STAT3の活性化を阻害することが示唆された(図2).
TRAF5はRINGドメインをもつことからユビキチンリガーゼとしてシグナル伝達に関与することが示唆された.しかし,さきに述べた結果は,TRAF5によるSTAT3の活性化の抑制にはRINGドメインが不要であることを意味した.そこで,TRAF5欠損CD4陽性T細胞にTRAF5欠失変異体を遺伝子導入しTh17細胞への分化について評価した.その結果,TRAFドメインを含むTRAF5欠失変異体は,野生型のTRAF5と同じ程度でTh17細胞への分化を抑制したが,RINGドメインを含むTRAF5欠失変異体にはTh17細胞への分化に対する抑制能は認められなかった.以上から,TRAF5によるTh17細胞への分化の抑制にはTRAFドメインが必要であることがわかった.
そこで,CD4陽性T細胞におけるTRAF5とgp130との相互作用がTh17細胞への分化の抑制にどのように重要かについて調べた.gp130のTRAF5結合領域を培養細胞において発現させることにより,TRAF5とgp130とのあいだの結合を阻害できることがわかった.したがって,この部分ペプチドを発現させることにより内在性のTRAF5とgp130との結合が阻害され,これによりTRAF5のインターロイキン6シグナルに対する抑制作用を解除できることが期待された.GFPのC末端側にgp130のTRAF5結合領域を付加した融合タンパク質をCD4陽性T細胞において発現させ,Th17細胞への分化にどのような影響を及ぼすかを調べた.その結果,野生型のCD4陽性T細胞にこの融合タンパク質を発現させることによりTh17細胞への分化は亢進したが,TRAF5欠損CD4陽性T細胞においてはこのような効果は確認できなかった.以上の結果から,TRAF5のgp130への結合こそがTh17細胞への分化の抑制に重要であることが強く示唆された.
その発症にTh17細胞が重要な役割をはたすことが知られている実験的自己免疫性脳脊髄炎(experimental autoimmune encephalomyelitis)を用いて,TRAF5が病態の形成にどのような影響を及ぼすかについて評価した.野生型マウスおよびTRAF5ノックアウトマウスをミエリンオリゴデンドロサイト糖タンパク質に由来する抗原ペプチドおよび完全フロイントアジュバントにより免疫し,8日後に所属リンパ節の細胞を調製し,抗原ペプチドにより再刺激したときに産生された培養上清におけるサイトカインの濃度を測定した.TRAF5ノックアウトマウスに由来するリンパ節の細胞の培養液からより高濃度のインターロイキン17およびインターフェロンγが検出された.さらに,TRAF5ノックアウトマウスにおいて実験的自己免疫性脳脊髄炎の病態の有意な増悪が観察され,これに対応して,中枢神経組織におけるインターフェロンγ陽性T細胞の数およびインターロイキン17陽性CD4陽性T細胞の数の有意な増加が認められた.実験的自己免疫性脳脊髄炎においてミエリンオリゴデンドロサイト糖タンパク質に特異的なインターフェロンγ陽性細胞はインターロイキン17陽性細胞の再分化により生じることが明らかになっており3,4),TRAF5ノックアウトマウスにおけるインターフェロンγ陽性細胞の増加はTh17細胞の分化の亢進に起因することが推察された.また,野生型マウスおよびTRAF5ノックアウトマウスから精製したCD4陽性T細胞を放射線を照射した野生型マウスに養子移入することにより実験的自己免疫性脳脊髄炎を誘導した結果,TRAF5欠損CD4陽性T細胞を移入したマウスにおいて有意な病態の亢進が認められた.この結果から,CD4陽性T細胞におけるTRAF5の欠損が実験的自己免疫性脳脊髄炎の増悪に結びつくことがわかった.以上の結果から,CD4陽性T細胞においてTRAF5が炎症性CD4陽性T細胞への分化を抑制し,その結果,自己免疫疾患の発症を制限する機構が明らかになった.
今回,筆者らは,TRAF5がgp130と恒常的に結合することによりインターロイキン6の刺激に依存的なSTAT3の活性化を阻害し,これにより炎症性CD4陽性T細胞への分化を抑制する機構を明らかにした.従来,TRAF5はTNF受容体スーパーファミリーの関与するシグナル伝達においてNF-κBなどによる炎症性シグナルを促進する機能をもつことがわかっていた.筆者らの研究から,TRAF5がインターロイキン6シグナルを抑制することにより抗炎症的にはたらくという予想外のシグナル伝達機構がはじめて明らかになった.TRAF1,TRAF2,TRAF3,TRAF5のもつTRAFドメインは同様のアミノ酸配列を認識することから,TRAF5以外のTRAFファミリータンパク質もインターロイキン6シグナルの制御に関与している可能性がある.また,受容体としてgp130を共有するインターロイキン6以外のサイトカイン,CNTF,CT-1,インターロイキン11,インターロイキン27,LIF,OSMなどのシグナルをTRAF5が制御している可能性もある.今後,TRAFファミリータンパク質によるgp130シグナルの制御機構の普遍性についても明らかにしていきたい.
略歴:東北大学大学院医学系研究科博士課程 在学中.
研究テーマ:CD4陽性T細胞の制御におけるTRAF5の機能.
抱負:免疫系の恒常性がいかに厳密に維持され,どのようなときに異常をきたすのかを解き明かし,これをさまざまな免疫疾患の治療法の開発に役だてたい.
宗 孝紀(Takanori So)
東北大学大学院医学系研究科 准教授.
© 2014 長島宏行・宗 孝紀 Licensed under CC 表示 2.1 日本
(東北大学大学院医学系研究科 免疫学分野)
email:宗 孝紀
DOI: 10.7875/first.author.2014.046
The adaptor TRAF5 limits the differentiation of inflammatory CD4+ T cells by antagonizing signaling via the receptor for IL-6.
Hiroyuki Nagashima, Yuko Okuyama, Atsuko Asao, Takeshi Kawabe, Satoshi Yamaki, Hiroyasu Nakano, Michael Croft, Naoto Ishii, Takanori So
Nature Immunology, 15, 449-456 (2014)
要 約
アダプタータンパク質TRAF5はTNF受容体スーパーファミリーによる炎症性シグナル応答にかかわることが知られていたが,CD4陽性T細胞におけるその詳細な機能は不明であった.今回,筆者らは,TRAF5がCD4陽性T細胞の分化の過程においてインターロイキン6シグナルを抑制し,これがヘルパーT細胞のサブセットであるTh17細胞の分化に対し抑制的にはたらくという予想外のシグナル伝達機構を見い出した.すなわち,TRAF5がシグナル伝達タンパク質であるgp130と恒常的に結合することにより,インターロイキン6の刺激に依存して起こるSTAT3のgp130へのリクルートおよび活性化を阻害することを発見した.さらに,Th17細胞に依存的な自己免疫疾患モデルである実験的自己免疫性脳脊髄炎においては,TRAF5の欠損により症状が増悪することがわかった.以上より,TRAF5がインターロイキン6の刺激による炎症性ヘルパーT細胞の分化を抑制することで,自己免疫疾患の発症および病態を制限する機構が明らかにされた.
はじめに
CD4陽性ヘルパーT細胞はB細胞による抗体の産生を誘導し,マクロファージやCD8陽性T細胞を活性化する.その体液性免疫および細胞性免疫に対する活性化の作用は諸刃の剣であり,病原体やがん細胞の排除にかかわる一方で,アレルギーや自己免疫疾患の病態の形成と関連する.ヘルパーT細胞の分化および機能における制御機構の解明は免疫学における重要な課題であり,現在,ヘルパーT細胞を標的にした疾患の制御に関する応用研究も精力的に行われている.
外来性の抗原とまだ接触したことのないナイーブCD4陽性T細胞は3つの異質なシグナル,すなわち,T細胞受容体シグナル,補助刺激受容体シグナル,サイトカイン受容体シグナルを受け取ることによりヘルパーT細胞へと分化する(図1).とりわけ重要なことは,受け取るサイトカイン受容体シグナルの質的な差により機能の異なるヘルパーT細胞のサブセットへと個別に分化することである(図1).Th17細胞はインターロイキン17を産生することにより好中球の感染部位への動員を促進する作用をもつ一方で,多発性硬化症,リウマチ性関節炎,乾癬,強直性脊椎炎など自己免疫性疾患の病態との関連が示唆される炎症性ヘルパーT細胞のサブセットである.このTh17細胞の分化には炎症性サイトカインであるインターロイキン6によるサイトカイン受容体シグナルが必要であるが,その詳細な分化制御の機構にはいまだ不明な点が多い.
TRAF5(tumor necrosis factor receptor-associated factor 5)はTRAF1からTRAF6までの6つのファミリータンパク質のひとつで,N末端側からC末端側へRINGドメイン,ジンクフィンガードメイン,TRAFドメインから構成される.RINGドメインおよびTRAFドメインにはそれぞれユビキチンリガーゼ活性およびアダプタータンパク質としての活性が認められ,これらによりNF-κBなどによる炎症性シグナル応答が促進される.TRAF5ノックアウトマウスは発生や生育に関しては正常であり,リンパ器官の形成やリンパ球の成熟において顕著な異常はとくに認められない1).
筆者らは,この研究にいたるまえ,TRAF5ノックアウトマウスにおいて肺組織における炎症の病態が亢進し,これがTRAF5欠損CD4陽性T細胞の過剰な反応性に起因するという予想に反した結果を得ていた2).この研究では,CD4陽性T細胞におけるTRAF5の機能を解析し,TRAF5がインターロイキン6受容体からのシグナルに対し抑制作用をもち,これにより炎症性Th17細胞の分化を制限する機構がはじめて明らかにされた.
1.TRAF5はTh17細胞への分化を抑制する
CD4陽性T細胞におけるTRAF5の機能を調べるため,野生型マウスおよびTRAF5ノックアウトマウスからナイーブCD4陽性T細胞を精製し,抗CD3抗体および抗CD28抗体,および,各種のサイトカインの刺激により,Th1細胞サブセット,Th2細胞サブセット,Th17細胞サブセット,制御性T細胞サブセットへの分化をin vitroにおいて誘導した(図1).その結果,TRAF5欠損ナイーブCD4陽性T細胞からヘルパーT細胞への分化の過程において培養上清に高濃度のインターロイキン17およびインターロイキン21が検出され,最終的にTh17細胞への分化の亢進が認められた.この結果と一致して,Th17細胞に関連する遺伝子であるRorc遺伝子,Il17a遺伝子,Il17f遺伝子,Il23r遺伝子の発現がTRAF5欠損CD4陽性T細胞において有意に上昇することがわかった.さらに,ヘルパーT細胞への分化がより生理的に誘導されるナイーブCD4陽性T細胞と抗原提示細胞との共培養実験系においても,TRAF5欠損CD4陽性T細胞においてTh17細胞への分化の亢進が認められた.細胞増殖能,インターロイキン2の産生能,ほかのヘルパーT細胞サブセットへの分化に関して有意な差が認められなかったことから,TRAF5はTh17細胞への分化を特異的に抑制することが示唆された.
in vitroにおいて観察されたTh17細胞への分化における差が生体においても同様に観察されるかどうか調べるため,卵白アルブミン抗原に特異的なT細胞受容体を発現するCD4陽性T細胞を用いて養子移入実験を行った.野生型マウスおよびTRAF5ノックアウトマウスからナイーブCD4陽性T細胞を精製し,野生型マウスに移入し卵白アルブミンおよび完全フロイントアジュバントによりマウスを免疫したのち,1週間後に所属リンパ節におけるTh17細胞への分化について評価した.その結果,ドナーとなった細胞にしめるインターロイキン17産生細胞の割合および絶対数がTRAF5欠損CD4陽性細胞において有意に増加していたことから,生体におけるヘルパーT細胞への分化においてもTRAF5がTh17細胞への分化に対し抑制的にはたらくことが明らかになった.
2.TRAF5はTh17細胞への分化において必須のインターロイキン6シグナルを抑制する
Th17細胞はインターロイキン6およびTGFβの刺激により,制御性T細胞はTGFβの刺激により分化する(図1).制御性T細胞への分化あるいはTGFβシグナルに対するTRAF5の欠損の影響はほとんど認められなかったことから,TRAF5にインターロイキン6シグナルに対する制御能のあることが示唆された.インターロイキン6はインターロイキン6受容体(CD126)とgp130(CD130)から構成される受容体に結合することによりgp130の二量体化を促進し,gp130の細胞内領域に結合するキナーゼであるJAKを活性化し,gp130の細胞内領域のリン酸化を誘導する.STAT3はリン酸化したgp130と結合することによりリン酸化され,二量体化したのち核へと移行し転写因子としての機能を発揮する(図2).インターロイキン6シグナル伝達経路に対するTRAF5の機能を調べる目的で,野生型マウスおよびTRAF5ノックアウトマウスからCD4陽性T細胞を精製し,インターロイキン6の刺激により誘導されるSTAT3のリン酸化のレベルを比較した.その結果,TRAF5の欠損によりSTAT3のリン酸化が有意に亢進することが判明した.また,CD8陽性T細胞においても同様の結果が得られた.B細胞やマクロファージなどTRAF5あるいはgp130,または,その両方の発現レベルの低い細胞では,TRAF5のSTAT3リン酸化に対する抑制能はほとんど検出できなかった.また,インターロイキン6と同様にSTAT3のリン酸化を誘導するインターロイキン10やインターロイキン21によりCD4陽性T細胞を刺激しても,TRAF5の欠損による影響は認められなかった.これら一連の実験から,TRAF5がインターロイキン6受容体を介したSTAT3のリン酸化を特異的に抑制し,しかも,その効果は細胞種によらず,TRAF5とgp130の両者の発現量に依存することがわかった.
3.TRAF5はインターロイキン6受容体からのシグナル伝達タンパク質であるgp130に恒常的に結合する
TRAF5によるインターロイキン6シグナルの抑制機構を調べる目的で,培養細胞にgp130,STAT3,TRAF5を一過性に発現させて,この3つのタンパク質の相互作用を免疫沈降法により評価した.その結果,TRAF5がインターロイキン6の刺激によらず恒常的にgp130と結合することが判明した.また,TRAF5の存在下ではインターロイキン6の刺激に依存的に起こるSTAT3とgp130とのあいだの相互作用が阻害された.重要なことに,TRAF5とgp130とのあいだの恒常的な結合はマウスから精製したCD4陽性T細胞においても検出され,この結合が生理的な環境においても起こることがわかった.これらの実験から,TRAF5がgp130に結合することによりSTAT3のgp130へのリクルートを阻害し,その結果,STAT3の活性化が抑制されるという機構が明らかになった(図2).
TRAF5とgp130がそれぞれどの領域を介して結合するかを調べた.RINGドメインおよびジンクフィンガードメインを含むTRAF5欠失変異体,および,TRAFドメインを含むTRAF5欠失変異体を作製し,gp130との結合能について評価した.その結果,TRAFドメインを含むTRAF5欠失変異体にgp130との結合活性が認められた.gp130についても細胞内領域のさまざまな欠失変異体や部位特異的な変異体を作製しTRAF5との相互作用を調べた.その結果,TRAF5がgp130の細胞内領域のアミノ酸配列774~798に存在する2つの領域と結合することが判明した.この領域の近傍にはSTAT3との結合部位が存在したことから,TRAF5とgp130とのあいだの結合がSTAT3とgp130とのあいだの結合に対し立体障害的にはたらくことにより,STAT3の活性化を阻害することが示唆された(図2).
4.TRAF5はgp130と結合することによりTh17細胞への分化を抑制する
TRAF5はRINGドメインをもつことからユビキチンリガーゼとしてシグナル伝達に関与することが示唆された.しかし,さきに述べた結果は,TRAF5によるSTAT3の活性化の抑制にはRINGドメインが不要であることを意味した.そこで,TRAF5欠損CD4陽性T細胞にTRAF5欠失変異体を遺伝子導入しTh17細胞への分化について評価した.その結果,TRAFドメインを含むTRAF5欠失変異体は,野生型のTRAF5と同じ程度でTh17細胞への分化を抑制したが,RINGドメインを含むTRAF5欠失変異体にはTh17細胞への分化に対する抑制能は認められなかった.以上から,TRAF5によるTh17細胞への分化の抑制にはTRAFドメインが必要であることがわかった.
そこで,CD4陽性T細胞におけるTRAF5とgp130との相互作用がTh17細胞への分化の抑制にどのように重要かについて調べた.gp130のTRAF5結合領域を培養細胞において発現させることにより,TRAF5とgp130とのあいだの結合を阻害できることがわかった.したがって,この部分ペプチドを発現させることにより内在性のTRAF5とgp130との結合が阻害され,これによりTRAF5のインターロイキン6シグナルに対する抑制作用を解除できることが期待された.GFPのC末端側にgp130のTRAF5結合領域を付加した融合タンパク質をCD4陽性T細胞において発現させ,Th17細胞への分化にどのような影響を及ぼすかを調べた.その結果,野生型のCD4陽性T細胞にこの融合タンパク質を発現させることによりTh17細胞への分化は亢進したが,TRAF5欠損CD4陽性T細胞においてはこのような効果は確認できなかった.以上の結果から,TRAF5のgp130への結合こそがTh17細胞への分化の抑制に重要であることが強く示唆された.
5.TRAF5ノックアウトマウスではTh17細胞に依存的な実験的自己免疫性脳脊髄炎が増悪する
その発症にTh17細胞が重要な役割をはたすことが知られている実験的自己免疫性脳脊髄炎(experimental autoimmune encephalomyelitis)を用いて,TRAF5が病態の形成にどのような影響を及ぼすかについて評価した.野生型マウスおよびTRAF5ノックアウトマウスをミエリンオリゴデンドロサイト糖タンパク質に由来する抗原ペプチドおよび完全フロイントアジュバントにより免疫し,8日後に所属リンパ節の細胞を調製し,抗原ペプチドにより再刺激したときに産生された培養上清におけるサイトカインの濃度を測定した.TRAF5ノックアウトマウスに由来するリンパ節の細胞の培養液からより高濃度のインターロイキン17およびインターフェロンγが検出された.さらに,TRAF5ノックアウトマウスにおいて実験的自己免疫性脳脊髄炎の病態の有意な増悪が観察され,これに対応して,中枢神経組織におけるインターフェロンγ陽性T細胞の数およびインターロイキン17陽性CD4陽性T細胞の数の有意な増加が認められた.実験的自己免疫性脳脊髄炎においてミエリンオリゴデンドロサイト糖タンパク質に特異的なインターフェロンγ陽性細胞はインターロイキン17陽性細胞の再分化により生じることが明らかになっており3,4),TRAF5ノックアウトマウスにおけるインターフェロンγ陽性細胞の増加はTh17細胞の分化の亢進に起因することが推察された.また,野生型マウスおよびTRAF5ノックアウトマウスから精製したCD4陽性T細胞を放射線を照射した野生型マウスに養子移入することにより実験的自己免疫性脳脊髄炎を誘導した結果,TRAF5欠損CD4陽性T細胞を移入したマウスにおいて有意な病態の亢進が認められた.この結果から,CD4陽性T細胞におけるTRAF5の欠損が実験的自己免疫性脳脊髄炎の増悪に結びつくことがわかった.以上の結果から,CD4陽性T細胞においてTRAF5が炎症性CD4陽性T細胞への分化を抑制し,その結果,自己免疫疾患の発症を制限する機構が明らかになった.
おわりに
今回,筆者らは,TRAF5がgp130と恒常的に結合することによりインターロイキン6の刺激に依存的なSTAT3の活性化を阻害し,これにより炎症性CD4陽性T細胞への分化を抑制する機構を明らかにした.従来,TRAF5はTNF受容体スーパーファミリーの関与するシグナル伝達においてNF-κBなどによる炎症性シグナルを促進する機能をもつことがわかっていた.筆者らの研究から,TRAF5がインターロイキン6シグナルを抑制することにより抗炎症的にはたらくという予想外のシグナル伝達機構がはじめて明らかになった.TRAF1,TRAF2,TRAF3,TRAF5のもつTRAFドメインは同様のアミノ酸配列を認識することから,TRAF5以外のTRAFファミリータンパク質もインターロイキン6シグナルの制御に関与している可能性がある.また,受容体としてgp130を共有するインターロイキン6以外のサイトカイン,CNTF,CT-1,インターロイキン11,インターロイキン27,LIF,OSMなどのシグナルをTRAF5が制御している可能性もある.今後,TRAFファミリータンパク質によるgp130シグナルの制御機構の普遍性についても明らかにしていきたい.
文 献
- Nakano, H., Sakon, S., Koseki, H. et al.: Targeted disruption of Traf5 gene causes defects in CD40- and CD27-mediated lymphocyte activation. Proc. Natl. Acad. Sci, USA, 96, 9803-9808 (1999)[PubMed]
- So, T., Salek-Ardakani, S., Nakano, H. et al.: TNF receptor-associated factor 5 limits the induction of Th2 immune responses. J. Immunol., 172, 4292-4297 (2004)[PubMed]
- Hirota, K., Duarte, J. H., Veldhoen, M. et al.: Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol., 12, 255-263 (2011)[PubMed]
- Serada, S., Fujimoto, M., Mihara, M. et al.: IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA, 105, 9041-9046 (2008)[PubMed]
著者プロフィール
略歴:東北大学大学院医学系研究科博士課程 在学中.
研究テーマ:CD4陽性T細胞の制御におけるTRAF5の機能.
抱負:免疫系の恒常性がいかに厳密に維持され,どのようなときに異常をきたすのかを解き明かし,これをさまざまな免疫疾患の治療法の開発に役だてたい.
宗 孝紀(Takanori So)
東北大学大学院医学系研究科 准教授.
© 2014 長島宏行・宗 孝紀 Licensed under CC 表示 2.1 日本
