iPS細胞における環状染色体の自己修復
林 洋平・山中伸弥
(米国Gladstone Institute of Cardiovascular Disease)
email:林 洋平,山中伸弥
DOI: 10.7875/first.author.2014.026
Cell-autonomous correction of ring chromosomes in human induced pluripotent stem cells.
Marina Bershteyn, Yohei Hayashi, Guillaume Desachy, Edward C. Hsiao, Salma Sami, Kathryn M. Tsang, Lauren A. Weiss, Arnold R. Kriegstein, Shinya Yamanaka, Anthony Wynshaw-Boris
Nature, 507, 99-103 (2014)
環状染色体は,一般的に同一の染色体の長腕および短腕の末端が欠失をともない融合することにより形成される.先天的な環状染色体の形成はさまざまな発育異常や精神遅滞に関与することが知られている.しかし,ほかの染色体異常と同様に,これらの疾患に対する根本的な治療法は存在しない.また,脊椎動物において環状染色体を再現性よく形成できるモデルがないため,その挙動や機構については未解明な点が多い.そこで,筆者らは,環状染色体をもつ患者からiPS細胞を作製した.解析の結果,予想外にも,樹立されたiPS細胞株のほとんどは環状染色体を失い,染色体の不分離により片親性ダイソミーへと変化していることが見い出された.これらの結果は,多能性幹細胞の維持における染色体分配の制御について新たな知見をもたらすものであり,iPS細胞を用いた新しい“染色体治療”の可能性を示唆した.
環状染色体は1926年にショウジョウバエにおいて1),ついで,1938年にトウモロコシにおいて発見された2),古典的な染色体異常の例として知られており,一般的に同一の染色体の長腕および短腕の末端が欠失をともない融合することにより形成される.ヒトでは1962年から環状染色体の臨床例があいついで報告され,これまでに先天的な疾患やがんへの関与が知られている.これらの病態について個々の患者の症状は千差万別であり,一般的な“環状染色体症候群”としては定義されていない3).その理由として,環状染色体はおのおのの染色体において生じうること,個々の環状染色体の形成にともなう欠失の大きさあるいは部位は異なること,一般的に環状染色体は細胞分裂の際に不安定だがその不安定さは個々の細胞株により異なること,さらに,非常にまれであり出現頻度の推定がむずかしいこと,などがあげられる.現在でも,ほかの染色体異常によりひき起こされる疾患と同様に,環状染色体そのものにアプローチする根本的な治療法はなく,患者ごとに対症療法が実施されている.
環状染色体の動態あるいは形成機構を解明するためのモデル系としては,さきのトウモロコシやショウジョウバエのようなモデル生物を利用したものがあげられる.また,放射線や紫外線の照射により環状染色体の形成が誘発されることが知られている.しかし,脊椎動物において安定した環状染色体をもつ細胞モデルは知られておらず,環状染色体がもたらす病態の解明につながるような分子生物学的な知見には不明な点が多かった.そこで,筆者らは,ヒトの細胞における環状染色体の動態とそれがもたらす病態のモデルを開発するため,環状染色体をともなう疾患の患者に由来する繊維芽細胞からiPS細胞(induced pluripotent stem cell,人工多能性幹細胞)を作製した4).その結果,予想外にも,環状染色体を安定に保持したiPS細胞株は作製が困難であった代わり,ほとんどは環状染色体を失い,もう一方の正常な染色体が染色体の不分離により片親性ダイソミーへと変化したiPS細胞株が得られた.
Miller-Dieker症候群は第17染色体の短腕の末端の欠失に起因する,重篤な滑脳症がおもな症状の優性遺伝病である5).第17染色体の環状染色体をもつMiller-Dieker症候群の患者1人の繊維芽細胞,および,環状染色体は形成せず欠失のみをもつMiller-Dieker症候群の患者2人の繊維芽細胞からそれぞれiPS細胞を作製し,その特徴を比較した.iPS細胞を作製する方法としては,筆者らの研究室で開発した,エピソーマルプラスミドをエレクトロポレーションにより導入する方法を用いた6,7).環状染色体をもつ患者に由来するiPS細胞様の6個のコロニーをピックアップし,5回の継代の段階でその核型を解析した.その結果,6株のうち4株のほとんど(90%以上)の細胞において環状染色体は欠失し正常な核型を示した.また,DNAフィンガープリンティングの結果から,これらのiPS細胞株は環状染色体をもつ患者に由来することが確認された.これらのiPS細胞株は未分化様の形態を維持し順調に増殖した.さらに,これらのiPS細胞株が多能性をもつことを胚葉体とテラトーマそれぞれの形成による三胚葉分化誘導実験により確認した.また,iPS細胞の作製に用いたプラスミドDNAのゲノムへの挿入はみられなかった.残りの2株は半分以上の細胞において環状染色体を維持していたが,突発的に分化したり細胞死を起こしたりする細胞が多く,正常なiPS細胞株として維持できなかった.また,環状染色体は形成せず欠失のみをもつ患者に由来するiPS細胞株も正常に樹立することができた.核型の解析により,これらのiPS細胞株には第17染色体の短腕の末端の欠失が確認された.以上の結果から,第17環状染色体をもつ患者に由来するiPS細胞株において正常な核型がみられたのは,第17染色体の短腕の末端の欠失が理由ではなく,環状染色体の形成に起因すると推測された.
環状染色体をもつ患者に由来するiPS細胞株が正常な核型を示した理由についてさらに検討するため,SNPアレイを用いて解析した.その結果,環状染色体をもつ患者ならびに環状染色体は形成せず欠失のみをもつ患者の繊維芽細胞,および,環状染色体は形成せず欠失のみをもつ患者に由来するiPS細胞株において,第17染色体の短腕の末端の欠失が確認された.一方,環状染色体をもつ患者に由来し正常な核型を示したiPS細胞株では,この欠失は消失し,どの染色体も2コピーが保持されていた.また,おのおののSNPがヘテロ接合かホモ接合かどうかを解析したところ,環状染色体をもつ患者に由来するiPS細胞株は第17染色体のほぼすべてのSNPがホモ接合であることが示された(ほかの染色体では,繊維芽細胞と同じように,ヘテロ接合とホモ接合が混在していた).以上の結果は,環状染色体をもつ患者に由来する正常な核型を示すiPS細胞株は,環状染色体ではないもう片方の完全な第17染色体が染色体の不分離により重複した片親性ダイソミーであることを示した.
この片親性ダイソミーの染色体が両方とも機能しているかどうか検討した.Miller-Dieker症候群において欠失している染色体の領域にはLIS1遺伝子および14-3-3e遺伝子があり,その病態において重要であることが知られている.これらの遺伝子のDNA量およびタンパク質の発現量をそれぞれの細胞について比較した.環状染色体をもつ患者の繊維芽細胞は,ほかの患者に由来する繊維芽細胞と同様に,これら遺伝子のDNA量およびタンパク質の発現量は野生型の繊維芽細胞と比較してほぼ半減していた.一方,環状染色体をもつ患者に由来する正常な核型を示したiPS細胞株では,これらの量が野生型のiPS細胞と同じ程度にまで回復していた.環状染色体は形成せず欠失のみをもつ患者に由来するiPS細胞でも,これらの量はほぼ半減していた.以上の結果は,環状染色体をもつ患者に由来するiPS細胞株において,片親性ダイソミーとなった2つの染色体それぞれから正常にタンパク質が発現されていることを示した.
第17染色体について見い出された結果がほかの染色体での環状染色体の形成においてもみられるかどうか検討するため,第13染色体の環状染色体をもつ2例の繊維芽細胞について解析した.これらの第13染色体の環状染色体はそれぞれ別の領域の欠失をともない,さまざまな組織の形成不全をともなう成長遅延ひき起こしていた.
これらの繊維芽細胞は80%以上の頻度で環状染色体を保持しており,この段階で正常な核型を示す細胞はみられなかった.これらの繊維芽細胞からiPS細胞を作製しその核型を同定した.その結果,継代数が8以上でほとんどすべての細胞が正常な核型を示す株がみられた.その一方,ある株では継代数が12になっても半分以上の細胞が環状染色体を保持していた.興味深いことに,別のある株では継代数が6の時点では90%以上の細胞が環状染色体を保持していたが,継代数が12の時点ではそのほとんどが正常な核型に置き換わっていた.DNAフィンガープリンティングの結果から,これらすべての試料は第13染色体の環状染色体を含む細胞に由来することが確認された.
これらの細胞株をSNPアレイを用いて解析したところ,繊維芽細胞では環状染色体の形成にともなう第13染色体の欠失が確認された.一方,これらの繊維芽細胞に由来するiPS細胞株,とくに正常な核型を示す細胞が大半のものでは,これらの欠失は消失していた.さらに,第13染色体のおのおののSNPがヘテロ接合かホモ接合かどうかを解析したところ,これらのiPS細胞では繊維芽細胞においてみられたヘテロ接合がホモ接合に置き換わっていた.以上の結果から,これらのiPS細胞株は片親性ダイソミーの第13染色体を保持していることが示された.また,これらのiPS細胞株は胚葉体とテラトーマそれぞれの形成による三胚葉分化誘導実験により多能性をもつことが確認された.さらにiPS細胞の作製に用いたプラスミドDNAのゲノムへの挿入はみられなかった.
以上の結果から,第17染色体の環状染色体のみならず,第13染色体の環状染色体をともなう細胞から樹立された大半のiPS細胞株においても,環状染色体の消失と片親性ダイソミーへの変化が観察された.今後の展望として,この結果がさらにほかの染色体における環状染色体においてもみられるかどうか,検証を重ねることは興味深い.
今回の研究により,第17染色体あるいは第13染色体の環状染色体をもつ繊維芽細胞から高頻度で片親性ダイソミーのiPS細胞が得られることを見い出した(図1).このような結果が得られた理由を,以上で得られた実験結果から考察した.
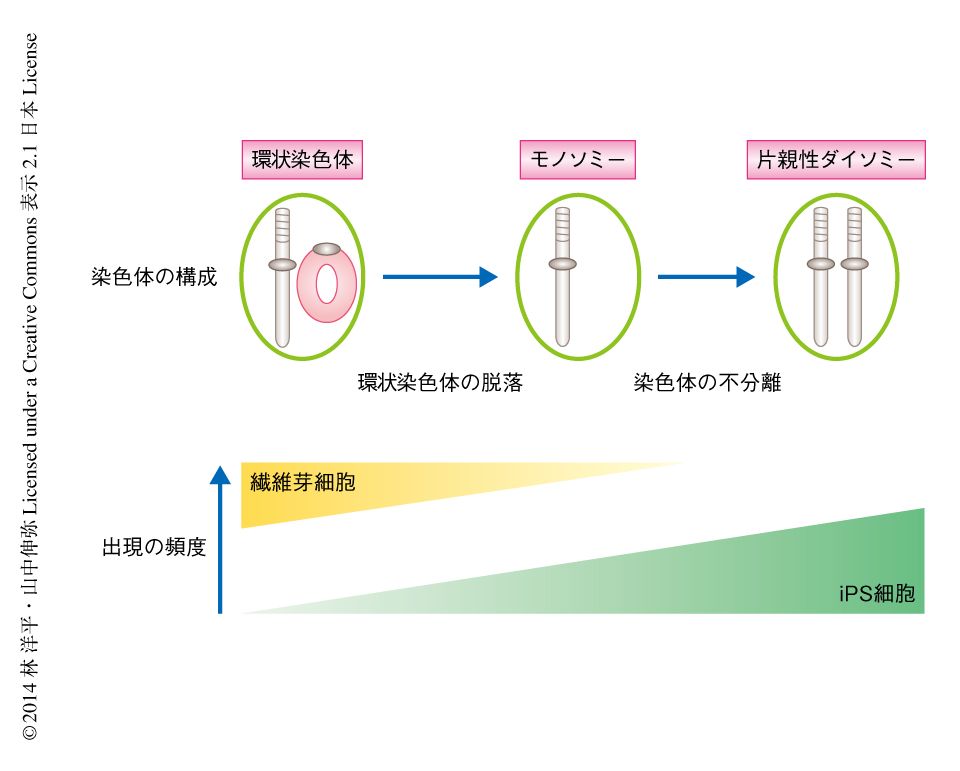
まず,どのような染色体の構成をへて片親性ダイソミーの細胞が得られたのかを考えた.その過程において,環状染色体の脱落が起こる,もう一方の完全な染色体が染色体の不分離により重複する,という2つのステップがそれぞれ独立して起こったことが想定された.そこで,それらのステップが起こった順序および時期を推定した.今回の核型の解析では,繊維芽細胞あるいはiPS細胞の一部に環状染色体およびその相同染色体がすべて脱落した細胞が出現したのに対して,環状染色体およびその2つの相同染色体をもつ細胞はほとんどみられなかった.また,DNA蛍光in situハイブリダイゼーション法においても,このような細胞はほぼみられなかった.以上の結果から,片親性ダイソミーにいたる中間状態として,繊維芽細胞,または,iPS細胞へのリプログラミングの途中で,環状染色体が脱落したモノソミーの細胞が出現したと考えられた.
つぎに,どのようにして大半の細胞が片親性ダイソミーの細胞になったのかを考えた.モノソミーから片親性ダイソミーにいたる様式として,染色体の不分離がまれな頻度で起こったことが想定された.具体的な頻度は明らかではないが,結果的に片親性ダイソミーの細胞が大半をしめていたことから,培養条件においてなんらかの淘汰圧が存在していたと考えられた.今回の核型の解析およびDNA蛍光in situハイブリダイゼーション法の結果から,第17染色体の片親性ダイソミーの細胞が大半をしめるiPS細胞株において,分裂中期の細胞を対象とした核型の解析ではほぼすべての細胞が片親性ダイソミーを示していたのに対し,すべての細胞を対象としたDNA蛍光in situハイブリダイゼーション法では一部の細胞において環状染色体をもつ細胞やモノソミーの細胞の残存が示唆された.この結果から,片親性ダイソミーの細胞は環状染色体をもつ細胞やモノソミーの細胞よりも分裂がさかんであり(生存および増殖に有利であり),培養条件において優勢となったと考えられた.
今後の研究では,これらの過程の機構を分子的に解明することが望まれる.
今回の研究では,環状染色体をともなう疾患の患者に由来するiPS細胞では,高頻度に環状染色体が消失し片親性ダイソミーへと変化するという結果を得た.この結果は,多能性幹細胞やヒトの発生初期における染色体分配の制御機構について新しい知見をもたらすと考えられる.これまで,ヒト多能性幹細胞8) やヒトの初期胚の細胞9) における染色体数の維持に対する不安定性が報告されている.今回の結果は,この不安定性を逆手にとるかたちで,正常な核型を示すiPS細胞が高頻度に得られたと考えられた.
この結果の臨床面への応用として,染色体異常をもつ患者からiPS細胞を作製し,その過程で染色体異常を片親性ダイソミーとして修正して,さらに再生移植医療を実施することが考えられる.もっとも,片親性ダイソミーには両親から受け継いだ正常な1対の染色体と比較して,大きく2つのリスクをかかえている10).まずひとつは,インプリンティング遺伝子の異常である.体細胞におけるインプリンティング遺伝子の発現の制御は父親に由来する染色体と母親に由来する染色体とでそれぞれ異なっているが,片親性ダイソミーでは両方の染色体においてその制御が同一となってしまうため,組織の分化や維持に異常をきたす場合がある.もうひとつは,ヘテロ接合では表現型に現われないような劣性変異が片親性ダイソミーでは顕在化する可能性である.今後の研究により,これらのリスクをiPS細胞からの分化誘導系や組織移植系において評価することが重要であると考えられる.以上の課題をクリアできた場合(さらに,もちろんiPS細胞からの再生移植医療がうまくいくことが最重要だが),今回の研究は環状染色体というまれな染色体異常を出発点としたが,ほかの一般的な染色体異常についても,異常をもつ染色体を,たとえば環状染色体の形成をへるなどして脱落させ,そこから片親性ダイソミーのiPS細胞株を作製することにより,“染色体治療”を含めたiPS細胞からの再生移植医療が展開できるのではないかと期待される.このような方法は,実現のためには技術的な課題も多いが,現在のゲノム編集技術では対応できない大規模な染色体異常に対し有効な治療法になりうるのではないかと考えられる.
略歴:2009年 東京大学大学院総合文化研究科博士課程 修了,同年より米国Gladstone Institute of Cardiovascular Disease博士研究員.
研究テーマ:稀少な疾患に特異的なiPS細胞およびそれを用いた疾患モデルの作製.
抱負:稀少な疾患の同定からiPS細胞の病態モデル,そして,創薬あるいは治療法の開発へと,“一気通貫”の研究を完成させたい.
山中 伸弥(Shinya Yamanaka)
京都大学iPS細胞研究所 所長.米国Gladstone Institute of Cardiovascular DiseaseにてSenior Investigator併任.
研究室URL:http://labs.gladstone.ucsf.edu/yamanaka
© 2014 林 洋平・山中伸弥 Licensed under CC 表示 2.1 日本
(米国Gladstone Institute of Cardiovascular Disease)
email:林 洋平,山中伸弥
DOI: 10.7875/first.author.2014.026
Cell-autonomous correction of ring chromosomes in human induced pluripotent stem cells.
Marina Bershteyn, Yohei Hayashi, Guillaume Desachy, Edward C. Hsiao, Salma Sami, Kathryn M. Tsang, Lauren A. Weiss, Arnold R. Kriegstein, Shinya Yamanaka, Anthony Wynshaw-Boris
Nature, 507, 99-103 (2014)
要 約
環状染色体は,一般的に同一の染色体の長腕および短腕の末端が欠失をともない融合することにより形成される.先天的な環状染色体の形成はさまざまな発育異常や精神遅滞に関与することが知られている.しかし,ほかの染色体異常と同様に,これらの疾患に対する根本的な治療法は存在しない.また,脊椎動物において環状染色体を再現性よく形成できるモデルがないため,その挙動や機構については未解明な点が多い.そこで,筆者らは,環状染色体をもつ患者からiPS細胞を作製した.解析の結果,予想外にも,樹立されたiPS細胞株のほとんどは環状染色体を失い,染色体の不分離により片親性ダイソミーへと変化していることが見い出された.これらの結果は,多能性幹細胞の維持における染色体分配の制御について新たな知見をもたらすものであり,iPS細胞を用いた新しい“染色体治療”の可能性を示唆した.
はじめに
環状染色体は1926年にショウジョウバエにおいて1),ついで,1938年にトウモロコシにおいて発見された2),古典的な染色体異常の例として知られており,一般的に同一の染色体の長腕および短腕の末端が欠失をともない融合することにより形成される.ヒトでは1962年から環状染色体の臨床例があいついで報告され,これまでに先天的な疾患やがんへの関与が知られている.これらの病態について個々の患者の症状は千差万別であり,一般的な“環状染色体症候群”としては定義されていない3).その理由として,環状染色体はおのおのの染色体において生じうること,個々の環状染色体の形成にともなう欠失の大きさあるいは部位は異なること,一般的に環状染色体は細胞分裂の際に不安定だがその不安定さは個々の細胞株により異なること,さらに,非常にまれであり出現頻度の推定がむずかしいこと,などがあげられる.現在でも,ほかの染色体異常によりひき起こされる疾患と同様に,環状染色体そのものにアプローチする根本的な治療法はなく,患者ごとに対症療法が実施されている.
環状染色体の動態あるいは形成機構を解明するためのモデル系としては,さきのトウモロコシやショウジョウバエのようなモデル生物を利用したものがあげられる.また,放射線や紫外線の照射により環状染色体の形成が誘発されることが知られている.しかし,脊椎動物において安定した環状染色体をもつ細胞モデルは知られておらず,環状染色体がもたらす病態の解明につながるような分子生物学的な知見には不明な点が多かった.そこで,筆者らは,ヒトの細胞における環状染色体の動態とそれがもたらす病態のモデルを開発するため,環状染色体をともなう疾患の患者に由来する繊維芽細胞からiPS細胞(induced pluripotent stem cell,人工多能性幹細胞)を作製した4).その結果,予想外にも,環状染色体を安定に保持したiPS細胞株は作製が困難であった代わり,ほとんどは環状染色体を失い,もう一方の正常な染色体が染色体の不分離により片親性ダイソミーへと変化したiPS細胞株が得られた.
1.第17染色体の環状染色体をもつ繊維芽細胞からの片親性ダイソミーのiPS細胞の作製
Miller-Dieker症候群は第17染色体の短腕の末端の欠失に起因する,重篤な滑脳症がおもな症状の優性遺伝病である5).第17染色体の環状染色体をもつMiller-Dieker症候群の患者1人の繊維芽細胞,および,環状染色体は形成せず欠失のみをもつMiller-Dieker症候群の患者2人の繊維芽細胞からそれぞれiPS細胞を作製し,その特徴を比較した.iPS細胞を作製する方法としては,筆者らの研究室で開発した,エピソーマルプラスミドをエレクトロポレーションにより導入する方法を用いた6,7).環状染色体をもつ患者に由来するiPS細胞様の6個のコロニーをピックアップし,5回の継代の段階でその核型を解析した.その結果,6株のうち4株のほとんど(90%以上)の細胞において環状染色体は欠失し正常な核型を示した.また,DNAフィンガープリンティングの結果から,これらのiPS細胞株は環状染色体をもつ患者に由来することが確認された.これらのiPS細胞株は未分化様の形態を維持し順調に増殖した.さらに,これらのiPS細胞株が多能性をもつことを胚葉体とテラトーマそれぞれの形成による三胚葉分化誘導実験により確認した.また,iPS細胞の作製に用いたプラスミドDNAのゲノムへの挿入はみられなかった.残りの2株は半分以上の細胞において環状染色体を維持していたが,突発的に分化したり細胞死を起こしたりする細胞が多く,正常なiPS細胞株として維持できなかった.また,環状染色体は形成せず欠失のみをもつ患者に由来するiPS細胞株も正常に樹立することができた.核型の解析により,これらのiPS細胞株には第17染色体の短腕の末端の欠失が確認された.以上の結果から,第17環状染色体をもつ患者に由来するiPS細胞株において正常な核型がみられたのは,第17染色体の短腕の末端の欠失が理由ではなく,環状染色体の形成に起因すると推測された.
環状染色体をもつ患者に由来するiPS細胞株が正常な核型を示した理由についてさらに検討するため,SNPアレイを用いて解析した.その結果,環状染色体をもつ患者ならびに環状染色体は形成せず欠失のみをもつ患者の繊維芽細胞,および,環状染色体は形成せず欠失のみをもつ患者に由来するiPS細胞株において,第17染色体の短腕の末端の欠失が確認された.一方,環状染色体をもつ患者に由来し正常な核型を示したiPS細胞株では,この欠失は消失し,どの染色体も2コピーが保持されていた.また,おのおののSNPがヘテロ接合かホモ接合かどうかを解析したところ,環状染色体をもつ患者に由来するiPS細胞株は第17染色体のほぼすべてのSNPがホモ接合であることが示された(ほかの染色体では,繊維芽細胞と同じように,ヘテロ接合とホモ接合が混在していた).以上の結果は,環状染色体をもつ患者に由来する正常な核型を示すiPS細胞株は,環状染色体ではないもう片方の完全な第17染色体が染色体の不分離により重複した片親性ダイソミーであることを示した.
この片親性ダイソミーの染色体が両方とも機能しているかどうか検討した.Miller-Dieker症候群において欠失している染色体の領域にはLIS1遺伝子および14-3-3e遺伝子があり,その病態において重要であることが知られている.これらの遺伝子のDNA量およびタンパク質の発現量をそれぞれの細胞について比較した.環状染色体をもつ患者の繊維芽細胞は,ほかの患者に由来する繊維芽細胞と同様に,これら遺伝子のDNA量およびタンパク質の発現量は野生型の繊維芽細胞と比較してほぼ半減していた.一方,環状染色体をもつ患者に由来する正常な核型を示したiPS細胞株では,これらの量が野生型のiPS細胞と同じ程度にまで回復していた.環状染色体は形成せず欠失のみをもつ患者に由来するiPS細胞でも,これらの量はほぼ半減していた.以上の結果は,環状染色体をもつ患者に由来するiPS細胞株において,片親性ダイソミーとなった2つの染色体それぞれから正常にタンパク質が発現されていることを示した.
2.第13染色体の環状染色体をもつ繊維芽細胞からの片親性ダイソミーのiPS細胞の作製
第17染色体について見い出された結果がほかの染色体での環状染色体の形成においてもみられるかどうか検討するため,第13染色体の環状染色体をもつ2例の繊維芽細胞について解析した.これらの第13染色体の環状染色体はそれぞれ別の領域の欠失をともない,さまざまな組織の形成不全をともなう成長遅延ひき起こしていた.
これらの繊維芽細胞は80%以上の頻度で環状染色体を保持しており,この段階で正常な核型を示す細胞はみられなかった.これらの繊維芽細胞からiPS細胞を作製しその核型を同定した.その結果,継代数が8以上でほとんどすべての細胞が正常な核型を示す株がみられた.その一方,ある株では継代数が12になっても半分以上の細胞が環状染色体を保持していた.興味深いことに,別のある株では継代数が6の時点では90%以上の細胞が環状染色体を保持していたが,継代数が12の時点ではそのほとんどが正常な核型に置き換わっていた.DNAフィンガープリンティングの結果から,これらすべての試料は第13染色体の環状染色体を含む細胞に由来することが確認された.
これらの細胞株をSNPアレイを用いて解析したところ,繊維芽細胞では環状染色体の形成にともなう第13染色体の欠失が確認された.一方,これらの繊維芽細胞に由来するiPS細胞株,とくに正常な核型を示す細胞が大半のものでは,これらの欠失は消失していた.さらに,第13染色体のおのおののSNPがヘテロ接合かホモ接合かどうかを解析したところ,これらのiPS細胞では繊維芽細胞においてみられたヘテロ接合がホモ接合に置き換わっていた.以上の結果から,これらのiPS細胞株は片親性ダイソミーの第13染色体を保持していることが示された.また,これらのiPS細胞株は胚葉体とテラトーマそれぞれの形成による三胚葉分化誘導実験により多能性をもつことが確認された.さらにiPS細胞の作製に用いたプラスミドDNAのゲノムへの挿入はみられなかった.
以上の結果から,第17染色体の環状染色体のみならず,第13染色体の環状染色体をともなう細胞から樹立された大半のiPS細胞株においても,環状染色体の消失と片親性ダイソミーへの変化が観察された.今後の展望として,この結果がさらにほかの染色体における環状染色体においてもみられるかどうか,検証を重ねることは興味深い.
3.片親性ダイソミーのiPS細胞株の出現に関する考察
今回の研究により,第17染色体あるいは第13染色体の環状染色体をもつ繊維芽細胞から高頻度で片親性ダイソミーのiPS細胞が得られることを見い出した(図1).このような結果が得られた理由を,以上で得られた実験結果から考察した.
まず,どのような染色体の構成をへて片親性ダイソミーの細胞が得られたのかを考えた.その過程において,環状染色体の脱落が起こる,もう一方の完全な染色体が染色体の不分離により重複する,という2つのステップがそれぞれ独立して起こったことが想定された.そこで,それらのステップが起こった順序および時期を推定した.今回の核型の解析では,繊維芽細胞あるいはiPS細胞の一部に環状染色体およびその相同染色体がすべて脱落した細胞が出現したのに対して,環状染色体およびその2つの相同染色体をもつ細胞はほとんどみられなかった.また,DNA蛍光in situハイブリダイゼーション法においても,このような細胞はほぼみられなかった.以上の結果から,片親性ダイソミーにいたる中間状態として,繊維芽細胞,または,iPS細胞へのリプログラミングの途中で,環状染色体が脱落したモノソミーの細胞が出現したと考えられた.
つぎに,どのようにして大半の細胞が片親性ダイソミーの細胞になったのかを考えた.モノソミーから片親性ダイソミーにいたる様式として,染色体の不分離がまれな頻度で起こったことが想定された.具体的な頻度は明らかではないが,結果的に片親性ダイソミーの細胞が大半をしめていたことから,培養条件においてなんらかの淘汰圧が存在していたと考えられた.今回の核型の解析およびDNA蛍光in situハイブリダイゼーション法の結果から,第17染色体の片親性ダイソミーの細胞が大半をしめるiPS細胞株において,分裂中期の細胞を対象とした核型の解析ではほぼすべての細胞が片親性ダイソミーを示していたのに対し,すべての細胞を対象としたDNA蛍光in situハイブリダイゼーション法では一部の細胞において環状染色体をもつ細胞やモノソミーの細胞の残存が示唆された.この結果から,片親性ダイソミーの細胞は環状染色体をもつ細胞やモノソミーの細胞よりも分裂がさかんであり(生存および増殖に有利であり),培養条件において優勢となったと考えられた.
今後の研究では,これらの過程の機構を分子的に解明することが望まれる.
おわりに
今回の研究では,環状染色体をともなう疾患の患者に由来するiPS細胞では,高頻度に環状染色体が消失し片親性ダイソミーへと変化するという結果を得た.この結果は,多能性幹細胞やヒトの発生初期における染色体分配の制御機構について新しい知見をもたらすと考えられる.これまで,ヒト多能性幹細胞8) やヒトの初期胚の細胞9) における染色体数の維持に対する不安定性が報告されている.今回の結果は,この不安定性を逆手にとるかたちで,正常な核型を示すiPS細胞が高頻度に得られたと考えられた.
この結果の臨床面への応用として,染色体異常をもつ患者からiPS細胞を作製し,その過程で染色体異常を片親性ダイソミーとして修正して,さらに再生移植医療を実施することが考えられる.もっとも,片親性ダイソミーには両親から受け継いだ正常な1対の染色体と比較して,大きく2つのリスクをかかえている10).まずひとつは,インプリンティング遺伝子の異常である.体細胞におけるインプリンティング遺伝子の発現の制御は父親に由来する染色体と母親に由来する染色体とでそれぞれ異なっているが,片親性ダイソミーでは両方の染色体においてその制御が同一となってしまうため,組織の分化や維持に異常をきたす場合がある.もうひとつは,ヘテロ接合では表現型に現われないような劣性変異が片親性ダイソミーでは顕在化する可能性である.今後の研究により,これらのリスクをiPS細胞からの分化誘導系や組織移植系において評価することが重要であると考えられる.以上の課題をクリアできた場合(さらに,もちろんiPS細胞からの再生移植医療がうまくいくことが最重要だが),今回の研究は環状染色体というまれな染色体異常を出発点としたが,ほかの一般的な染色体異常についても,異常をもつ染色体を,たとえば環状染色体の形成をへるなどして脱落させ,そこから片親性ダイソミーのiPS細胞株を作製することにより,“染色体治療”を含めたiPS細胞からの再生移植医療が展開できるのではないかと期待される.このような方法は,実現のためには技術的な課題も多いが,現在のゲノム編集技術では対応できない大規模な染色体異常に対し有効な治療法になりうるのではないかと考えられる.
文 献
- Morgan, L. V.: Correlation between shape and behavior of a chromosome. Proc. Natl. Acad. Sci. USA, 12, 180-181 (1926)[PubMed]
- McClintock, B.: The production of homozygous deficient tissues with mutant characteristics by means of the aberrant mitotic behavior of ring-shaped chromosomes. Genetics, 23, 315-376 (1938)[PubMed]
- Kosztolanyi, G.: Does "ring syndrome" exist? An analysis of 207 case reports on patients with a ring autosome. Hum. Genet., 75, 174-179 (1987)[PubMed]
- Takahashi, K., Tanabe, K., Ohnuki, M. et al.: Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell, 131, 861-872 (2007)[PubMed]
- Wynshaw-Boris, A., Pramparo, T., Youn, Y. H. et al.: Lissencephaly: mechanistic insights from animal models and potential therapeutic strategies. Semin. Cell Dev. Biol., 21, 823-830 (2010)[PubMed]
- Okita, K., Matsumura, Y., Sato, Y. et al.: A more efficient method to generate integration-free human iPS cells. Nat. Methods, 8, 409-412 (2011)[PubMed]
- Matsumoto, Y., Hayashi, Y., Schlieve, C. R. et al.: Induced pluripotent stem cells from patients with human fibrodysplasia ossificans progressiva show increased mineralization and cartilage formation. Orphanet J. Rare Dis., 8, 190-204 (2013)[PubMed]
- Spits, C., Mateizel, I., Geens, M. et al.: Recurrent chromosomal abnormalities in human embryonic stem cells. Nat. Biotechnol., 26, 1361-1363 (2008)[PubMed]
- Vanneste, E., Voet, T., Caignec, C. L. et al.: Chromosome instability is common in human cleavage-stage embryos. Nat. Med., 15, 577-583 (2009)[PubMed]
- Robinson, W. P.: Mechanisms leading to uniparental disomy and their clinical consequences. Bioessays, 22, 452-459 (2000)[PubMed]
著者プロフィール
略歴:2009年 東京大学大学院総合文化研究科博士課程 修了,同年より米国Gladstone Institute of Cardiovascular Disease博士研究員.
研究テーマ:稀少な疾患に特異的なiPS細胞およびそれを用いた疾患モデルの作製.
抱負:稀少な疾患の同定からiPS細胞の病態モデル,そして,創薬あるいは治療法の開発へと,“一気通貫”の研究を完成させたい.
山中 伸弥(Shinya Yamanaka)
京都大学iPS細胞研究所 所長.米国Gladstone Institute of Cardiovascular DiseaseにてSenior Investigator併任.
研究室URL:http://labs.gladstone.ucsf.edu/yamanaka
© 2014 林 洋平・山中伸弥 Licensed under CC 表示 2.1 日本
