真核生物の転写開始複合体のクライオ電子顕微鏡による構造解析
村上 健次
(米国Stanford大学Department of Structural Biology)
email:村上健次
DOI: 10.7875/first.author.2013.144
Architecture of an RNA polymerase II transcription pre-initiation complex.
Kenji Murakami, Hans Elmlund, Nir Kalisman, David A. Bushnell, Christopher M. Adams, Maia Azubel, Dominika Elmlund, Yael Levi-Kalisman, Xin Liu, Brian J. Gibbons, Michael Levitt, Roger D. Kornberg
Science, 342, 1238724 (2013)
RNAポリメラーゼIIは転写反応においてRNA鎖を伸長することはできるが,遺伝子プロモーターを認識して転写を開始することはできない.これらの本質的な機能にはRNAポリメラーゼIIにくわえ,基本転写因子であるTFIIA,TFIIB,TFIID,TFIIE,TFIIF,TFIIHのセットが必要である.RNAポリメラーゼIIは基本転写因子によりプロモーター領域のDNAと結合し,2本鎖DNAをほどいて鋳型となるDNA鎖から転写を開始するが,これらの構造的な基盤についてはほとんどわかっていない.これまでに,基本転写因子のうちTFIIBのみがRNAポリメラーゼIIとの複合体としてX線結晶解析がなされていた.この論文では,RNAポリメラーゼII,基本転写因子,プロモーター領域のDNAから転写開始複合体を再構成することに成功し,クライオ電子顕微鏡による構造解析と化学架橋-質量分析法によりその構造的な基盤を明らかにした.
DNAはmRNAに転写されたのち,タンパク質に翻訳されることにより遺伝情報が細胞に伝えられる.受精卵においてはほとんどの遺伝子が転写されているが,受精卵から分化が進み決まった役割をもつようになった細胞では転写は制御され,それぞれの細胞において必要な遺伝子しか転写されないようになっている.また,遺伝子の発現はストレスなど外からの刺激,あるいは,老化などによっても影響をうける.現在,これらの遺伝子発現の情報を疾患の診断や治療に役だてる試みが広がりつつある.転写開始,転写伸長,転写終了,スプライシング,mRNAの核外輸送,クロマチン構造など,核における多様な分子機構が遺伝子発現の制御に寄与しているが,転写開始はそのなかでももっとも重要であると考えられる.
DNAからmRNAへの転写はRNAポリメラーゼIIにより行われる1-3).筆者らのグループは,15年をかけて出芽酵母のRNAポリメラーゼIIの立体構造を決定し,RNA鎖を“伸長”する分子機構を明らかにした4).しかし,転写の“開始”にはRNAポリメラーゼIIにくわえ,基本転写因子であるTFIIA,TFIIB,TFIID,TFIIE,TFIIF,TFIIHが必須である.RNAポリメラーゼIIと基本転写因子はすべてのmRNAの転写に必須で,転写開始ごとに“転写開始複合体”を形成する.また,多くの遺伝子の発現が転写開始複合体に多様な転写因子が結合することにより制御されている.
転写開始複合体の構造解析はこれまでに多くのグループにより取り組まれてきたが,タンパク質の精製のむずかしさ,また,試料の不安定さから,試料の調製にすらいたっていなかった.筆者らのグループは,20年以上をかけてこれらの問題に取り組んできたが,近年,ようやくすべての基本転写因子とRNAポリメラーゼIIを出芽酵母から高純度かつ高収量にて精製することに成功し,安定な転写開始複合体を再構成することができるようになった.これにより,電子顕微鏡解析,および,X線結晶解析といった構造学的な解析,あるいは,1分子解析など,in vitroにおけるほとんどすべての研究が可能になった.
この研究では,クライオ電子顕微鏡を用いた解析により転写開始複合体の立体構造を16Åの分解能で決定し,RNAポリメラーゼIIと基本転写因子がDNAのうえでどのように結合しているか,はじめて可視化することに成功した.
基本転写因子はその大きさや不安定性のため,大腸菌における発現や昆虫細胞などによる大量発現が困難である.とくにTFIIFとTFIIHについてはむずかしく,筆者らのグループも含めたすべての研究室は,長いあいだ,均一な試料を精製できずにいた.筆者らは,出芽酵母のゲノムにコードされる目的タンパク質にTAPタグを導入し,出芽酵母を100~200リットルと大量に培養することで,構造解析の可能なだけの収量を得ることをめざした.TFIIFは活性を保ちながら可溶化する変性剤を同定することにより精製が可能となり5),TFIIHはその不安定化タンパク質を同定しこの遺伝子をノックアウトすることにより均一な試料を精製できるようになった6).
高純度に精製された6種類の基本転写因子とRNAポリメラーゼIIを用いて転写開始複合体の再構成に取り組んだ.これらを高塩濃度で混ぜ合わせて透析するだけでは,すべて沈殿してしまい再構成はできなかった.基本転写因子をくわえる順序,塩濃度などのパラメーターを1年ほどかけて最適化したのち,たった1通りの方法で均一かつ高収量にて転写開始複合体を再構成できることがわかった5).この再構成法では多量の競合するDNAの存在のもとでも,TATA領域を含むわずか20~30 bp程度のプロモーター領域のうえに転写開始複合体を再構成し精製することが可能であった.再構成された転写開始複合体は高い転写活性を保持し,ヌクレオチドをくわえるとほぼ100%の効率でプロモーター領域の2本鎖DNAがほどけ,鋳型となる1本鎖DNAがRNAポリメラーゼIIのDNA結合クレフトに結合することも,1分子解析などから明らかになった.
クライオ電子顕微鏡により得られた画像の処理は,初期モデルを利用せず,単粒子解析により行った(EMDB ID:2394).転写開始複合体の密度はP-ローブとG-ローブの2つの部分に分かれており,プロモーター領域のDNAをP-ローブとG-ローブがサンドイッチするようなかたちになっていた(図1).P-ローブはX線結晶構造4) との類似からすぐにRNAポリメラーゼIIであることがわかった(図1a).RNAポリメラーゼIIとのドッキングは,電子顕微鏡により得られたP-ローブの密度に対しRNAポリメラーゼIIのX線結晶構造を手動であわせたのち,相関関数にもとづく自動ドッキングのプログラムを用いて最適化した.
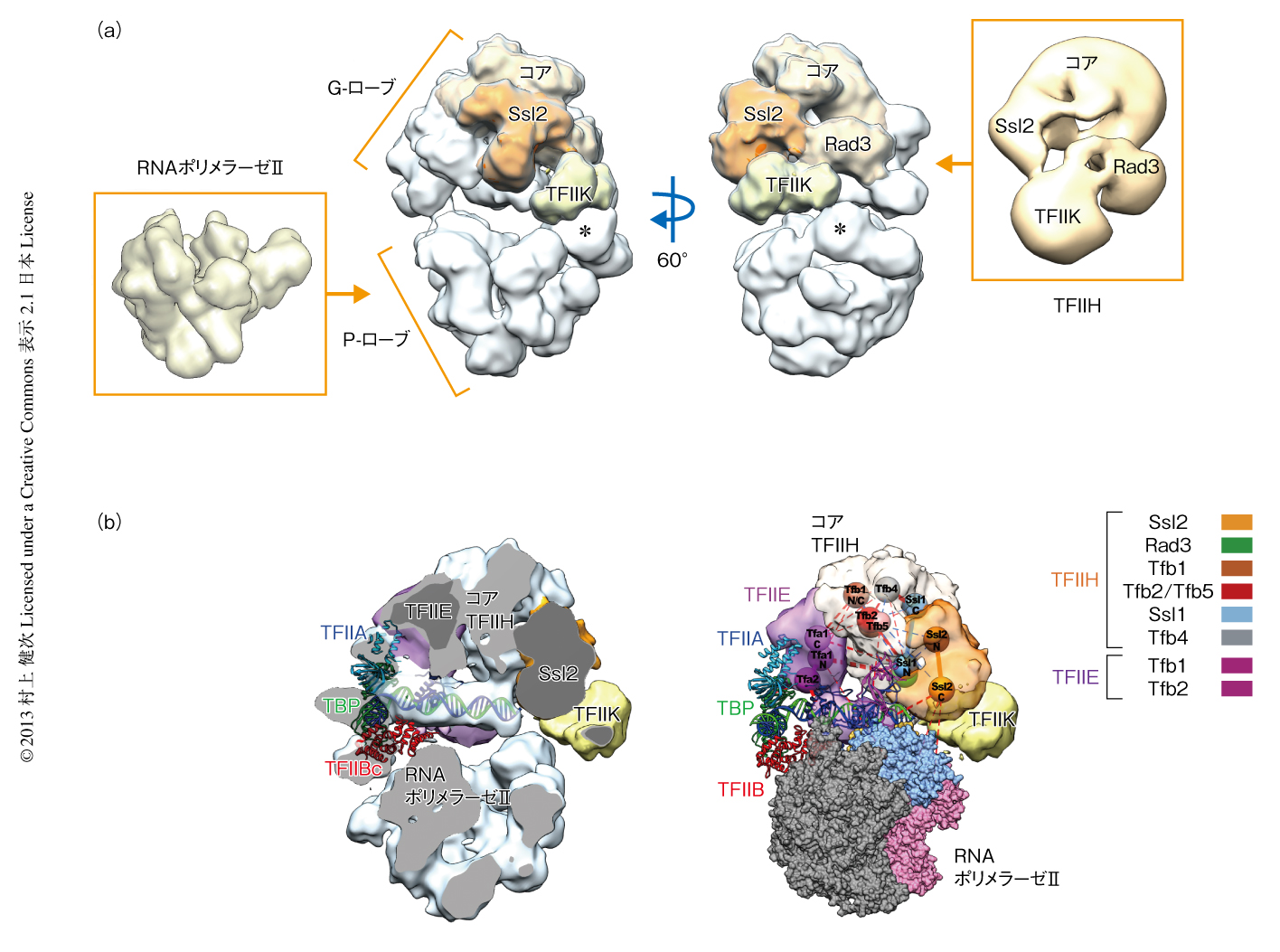
およそ1 MDaのG-ローブは,電子顕微鏡により得られたTFIIHの構造7) と大きさや形状などが類似していた(図1a).これにもとづき,TFIIHのもつ2つのヘリカーゼサブユニット(Rad3およびSsl2),および,リン酸化サブユニット(TFIIK)のおおよその位置を同定することができた.さらに,これらはTFIIKを除いて再構成をした転写開始複合体を電子顕微鏡により解析することで再確認することができた.
TFIIDの一部であるTBP,TFIIA,TFIIB,および,プロモーター領域のDNAに帰属される密度はRNAポリメラーゼII-TBP-TFIIB-DNA複合体のX線結晶構造のモデル8,9) とほぼ同一であった.このX線結晶構造によるモデルはDNAを15度ほど修正したのち,電子顕微鏡により得られた密度とぴったりドッキングすることができた(図1b).DNAの密度はG-ローブに強く結合しており,TATAボックスからSsl2まで円筒状の密度が確認された(図1b).
電子顕微鏡により得られた密度に対するドッキングは,分子量の大きな複合体を解析するには一般的な手法であるが,あくまで主観的であり客観的な同定が必要であった.そこで,化学架橋-質量分析法10) にもとづく自動帰属の方法を開発し適用することにより,すべての基本転写因子を同定することができた.この方法では,タンパク質において架橋の可能な距離(34Å未満)にあるリジン残基どうしをランダムに化学架橋し,プロテアーゼによりペプチドに切断し,架橋をもつペプチドのアミノ酸配列を質量分析により同定する.この方法により,およそ1.5 MDaの転写開始複合体から109個の分子間架橋と157個の分子内架橋を同定した.そのうち,73個の架橋は既知であるRNAポリメラーゼII-TFIIB複合体およびTFIIA-TBP-DNA複合体のX線結晶構造と一致したことから,データの妥当性が示された.
合計266個の架橋から得られた情報を電子顕微鏡により得られた密度に反映するため,自動帰属のプログラムを開発した.このプログラムにより,まず,分子内架橋にもとづき各サブユニットを1~3個の剛体,たとえば,Rad3のような球状タンパク質は1個の剛体,Ssl2は2個の剛体に分割した.つぎに,G-ローブを電子顕微鏡により得られた密度の分布にもとづき12個の剛体に分割した.この12個の剛体に対して12の階乗とおりの帰属がありうるが,これら12の階乗個のモデルおのおのに対し架橋のパターンとの適合を評価した.最終的に得られたモデルは266個の架橋のうち263個の架橋と一致し,一致しなかった3個の架橋は自由度の高いフレキシブルなループにあった.自動帰属の結果は電子顕微鏡により得られた密度に対するドッキングと完全に一致し(図1b),また,ほかのグループから得られていたDNA-タンパク質結合マッピング11) など,生化学的な実験の結果を非常によく説明できるものであった.
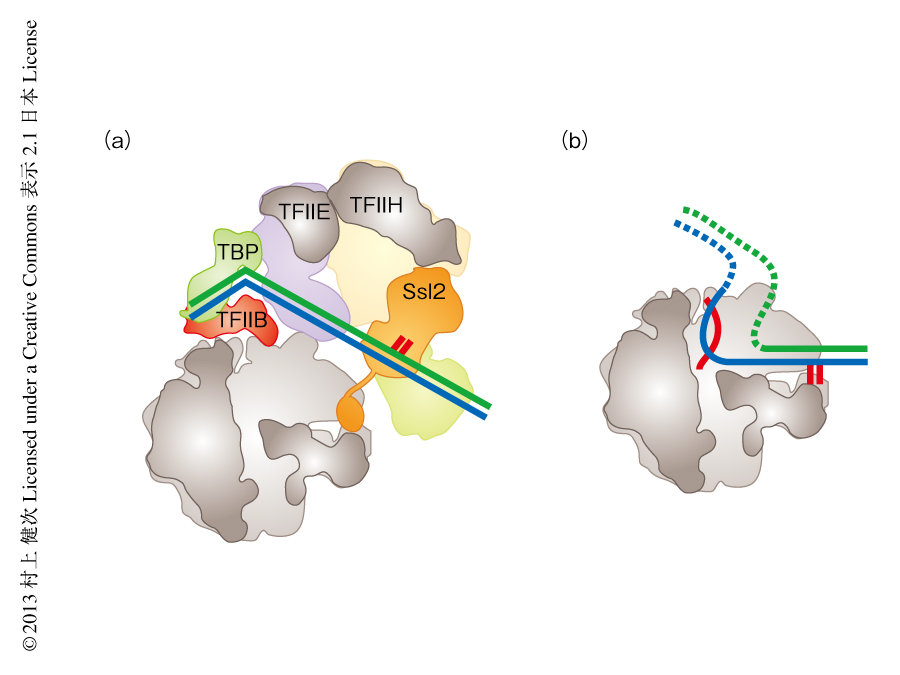
この研究により,基本転写因子とRNAポリメラーゼIIがプロモーター領域のDNAに対しどのように結合するかがはじめて明らかにされた.TFIIHのヘリカーゼサブユニットであるSsl2はTATAボックスから30Å以上も離れて位置しており,TATAボックスとSsl2のあいだのDNA領域はタンパク質との結合により安定化されず溶媒に露出していて,そのため,電子顕微鏡によるマップでは円筒状の密度として観察された.Ssl2のATPase活性による負のトルク力によりこの円筒状の密度の2本鎖DNAはほどかれ,鋳型となるDNA鎖はすぐそばにあるRNAポリメラーゼIIのDNA結合クレフトに結合できると考えられた.
プロモーター領域のDNAはRNAポリメラーゼIIのDNA結合クレフトのちょうど真上に位置していたが,RNAポリメラーゼIIとは直接には結合しておらずG-ローブに強く結合していた(図2).RNAポリメラーゼIIのDNA結合クレフトの内部にはSsl2のC末端領域が結合しており,DNAとの結合を阻害する機構が観察された.RNAポリメラーゼIIのDNA結合クレフトにはDNAがちょうど結合する空間しかないことから,RNAポリメラーゼIIが鋳型となるDNA鎖に結合したのち,Ssl2のC末端領域はRNAポリメラーゼIIのDNA結合クレフトから排除されると考えられた(図2).そののち,転写が開始され,転写開始複合体はいくつもの構造の変化を経由して転写伸長のステージに入るが,これらの構造解析は現在進行中である.
転写開始には基本転写因子とRNAポリメラーゼIIとが必要であることは20年以上もまえから知られていたが,それらが安定な転写開始複合体を形成していることは証明されていなかった.また,これまでのin vitroでの研究における転写活性は0.1~1.0%と本当にごくわずかで,放射能により標識したヌクレオチドを用いてようやく検知できるほどのものであった.筆者らの一連の研究においていちばん重要だったのは,転写開始複合体がゲルろ過カラムなどによる精製においても安定な複合体を保ち,クライオ電子顕微鏡やX線結晶回折により解析できるほどの安定性をもつことを証明して,高い転写活性をもつin vitro再構成系を確立したことである5,6).もちろん,転写開始複合体の構造解析も非常に困難であり,電子顕微鏡により得られた画像の処理法や化学架橋-質量分析法の技術改良は必須であった.
このようにして真核生物における転写開始はようやくin vitro再構成系が確立され,おそらく,つぎの10年から15年で転写の構造生物学はピークにいたると思われる.リボソームの構造が電子顕微鏡により30Åの分解能で解かれたのち,わずか10年たらずで原子構造までが解かれたことを考えれば,同じように転写開始複合体も原子構造が解明されることが期待できる.これらの研究はもちろん重要であるが,転写を研究する意義は“転写制御”の理解であることを忘れてはならないと思う.幸い,転写制御の分子機構の解明はまだ多くの地道な生化学的な研究が必要で,とくにin vitro再構成系においてはほとんどなにもされていない.1980年代から2000年代にかけて数人の研究者が転写の研究の土台を築いたように,今度は筆者の世代の研究者が転写制御の分野を切り開く必要があり,この分野はまだはじまったばかりである.
略歴:2004年 東京大学大学院理学系研究科 修了,2008年より米国Stanford大学 研究員.
研究テーマ:真核生物における転写制御.
© 2013 村上 健次 Licensed under CC 表示 2.1 日本
(米国Stanford大学Department of Structural Biology)
email:村上健次
DOI: 10.7875/first.author.2013.144
Architecture of an RNA polymerase II transcription pre-initiation complex.
Kenji Murakami, Hans Elmlund, Nir Kalisman, David A. Bushnell, Christopher M. Adams, Maia Azubel, Dominika Elmlund, Yael Levi-Kalisman, Xin Liu, Brian J. Gibbons, Michael Levitt, Roger D. Kornberg
Science, 342, 1238724 (2013)
要 約
RNAポリメラーゼIIは転写反応においてRNA鎖を伸長することはできるが,遺伝子プロモーターを認識して転写を開始することはできない.これらの本質的な機能にはRNAポリメラーゼIIにくわえ,基本転写因子であるTFIIA,TFIIB,TFIID,TFIIE,TFIIF,TFIIHのセットが必要である.RNAポリメラーゼIIは基本転写因子によりプロモーター領域のDNAと結合し,2本鎖DNAをほどいて鋳型となるDNA鎖から転写を開始するが,これらの構造的な基盤についてはほとんどわかっていない.これまでに,基本転写因子のうちTFIIBのみがRNAポリメラーゼIIとの複合体としてX線結晶解析がなされていた.この論文では,RNAポリメラーゼII,基本転写因子,プロモーター領域のDNAから転写開始複合体を再構成することに成功し,クライオ電子顕微鏡による構造解析と化学架橋-質量分析法によりその構造的な基盤を明らかにした.
はじめに
DNAはmRNAに転写されたのち,タンパク質に翻訳されることにより遺伝情報が細胞に伝えられる.受精卵においてはほとんどの遺伝子が転写されているが,受精卵から分化が進み決まった役割をもつようになった細胞では転写は制御され,それぞれの細胞において必要な遺伝子しか転写されないようになっている.また,遺伝子の発現はストレスなど外からの刺激,あるいは,老化などによっても影響をうける.現在,これらの遺伝子発現の情報を疾患の診断や治療に役だてる試みが広がりつつある.転写開始,転写伸長,転写終了,スプライシング,mRNAの核外輸送,クロマチン構造など,核における多様な分子機構が遺伝子発現の制御に寄与しているが,転写開始はそのなかでももっとも重要であると考えられる.
DNAからmRNAへの転写はRNAポリメラーゼIIにより行われる1-3).筆者らのグループは,15年をかけて出芽酵母のRNAポリメラーゼIIの立体構造を決定し,RNA鎖を“伸長”する分子機構を明らかにした4).しかし,転写の“開始”にはRNAポリメラーゼIIにくわえ,基本転写因子であるTFIIA,TFIIB,TFIID,TFIIE,TFIIF,TFIIHが必須である.RNAポリメラーゼIIと基本転写因子はすべてのmRNAの転写に必須で,転写開始ごとに“転写開始複合体”を形成する.また,多くの遺伝子の発現が転写開始複合体に多様な転写因子が結合することにより制御されている.
転写開始複合体の構造解析はこれまでに多くのグループにより取り組まれてきたが,タンパク質の精製のむずかしさ,また,試料の不安定さから,試料の調製にすらいたっていなかった.筆者らのグループは,20年以上をかけてこれらの問題に取り組んできたが,近年,ようやくすべての基本転写因子とRNAポリメラーゼIIを出芽酵母から高純度かつ高収量にて精製することに成功し,安定な転写開始複合体を再構成することができるようになった.これにより,電子顕微鏡解析,および,X線結晶解析といった構造学的な解析,あるいは,1分子解析など,in vitroにおけるほとんどすべての研究が可能になった.
この研究では,クライオ電子顕微鏡を用いた解析により転写開始複合体の立体構造を16Åの分解能で決定し,RNAポリメラーゼIIと基本転写因子がDNAのうえでどのように結合しているか,はじめて可視化することに成功した.
1.生化学的な解析
基本転写因子はその大きさや不安定性のため,大腸菌における発現や昆虫細胞などによる大量発現が困難である.とくにTFIIFとTFIIHについてはむずかしく,筆者らのグループも含めたすべての研究室は,長いあいだ,均一な試料を精製できずにいた.筆者らは,出芽酵母のゲノムにコードされる目的タンパク質にTAPタグを導入し,出芽酵母を100~200リットルと大量に培養することで,構造解析の可能なだけの収量を得ることをめざした.TFIIFは活性を保ちながら可溶化する変性剤を同定することにより精製が可能となり5),TFIIHはその不安定化タンパク質を同定しこの遺伝子をノックアウトすることにより均一な試料を精製できるようになった6).
高純度に精製された6種類の基本転写因子とRNAポリメラーゼIIを用いて転写開始複合体の再構成に取り組んだ.これらを高塩濃度で混ぜ合わせて透析するだけでは,すべて沈殿してしまい再構成はできなかった.基本転写因子をくわえる順序,塩濃度などのパラメーターを1年ほどかけて最適化したのち,たった1通りの方法で均一かつ高収量にて転写開始複合体を再構成できることがわかった5).この再構成法では多量の競合するDNAの存在のもとでも,TATA領域を含むわずか20~30 bp程度のプロモーター領域のうえに転写開始複合体を再構成し精製することが可能であった.再構成された転写開始複合体は高い転写活性を保持し,ヌクレオチドをくわえるとほぼ100%の効率でプロモーター領域の2本鎖DNAがほどけ,鋳型となる1本鎖DNAがRNAポリメラーゼIIのDNA結合クレフトに結合することも,1分子解析などから明らかになった.
2.電子顕微鏡による解析
クライオ電子顕微鏡により得られた画像の処理は,初期モデルを利用せず,単粒子解析により行った(EMDB ID:2394).転写開始複合体の密度はP-ローブとG-ローブの2つの部分に分かれており,プロモーター領域のDNAをP-ローブとG-ローブがサンドイッチするようなかたちになっていた(図1).P-ローブはX線結晶構造4) との類似からすぐにRNAポリメラーゼIIであることがわかった(図1a).RNAポリメラーゼIIとのドッキングは,電子顕微鏡により得られたP-ローブの密度に対しRNAポリメラーゼIIのX線結晶構造を手動であわせたのち,相関関数にもとづく自動ドッキングのプログラムを用いて最適化した.
およそ1 MDaのG-ローブは,電子顕微鏡により得られたTFIIHの構造7) と大きさや形状などが類似していた(図1a).これにもとづき,TFIIHのもつ2つのヘリカーゼサブユニット(Rad3およびSsl2),および,リン酸化サブユニット(TFIIK)のおおよその位置を同定することができた.さらに,これらはTFIIKを除いて再構成をした転写開始複合体を電子顕微鏡により解析することで再確認することができた.
TFIIDの一部であるTBP,TFIIA,TFIIB,および,プロモーター領域のDNAに帰属される密度はRNAポリメラーゼII-TBP-TFIIB-DNA複合体のX線結晶構造のモデル8,9) とほぼ同一であった.このX線結晶構造によるモデルはDNAを15度ほど修正したのち,電子顕微鏡により得られた密度とぴったりドッキングすることができた(図1b).DNAの密度はG-ローブに強く結合しており,TATAボックスからSsl2まで円筒状の密度が確認された(図1b).
3.化学架橋-質量分析法による解析
電子顕微鏡により得られた密度に対するドッキングは,分子量の大きな複合体を解析するには一般的な手法であるが,あくまで主観的であり客観的な同定が必要であった.そこで,化学架橋-質量分析法10) にもとづく自動帰属の方法を開発し適用することにより,すべての基本転写因子を同定することができた.この方法では,タンパク質において架橋の可能な距離(34Å未満)にあるリジン残基どうしをランダムに化学架橋し,プロテアーゼによりペプチドに切断し,架橋をもつペプチドのアミノ酸配列を質量分析により同定する.この方法により,およそ1.5 MDaの転写開始複合体から109個の分子間架橋と157個の分子内架橋を同定した.そのうち,73個の架橋は既知であるRNAポリメラーゼII-TFIIB複合体およびTFIIA-TBP-DNA複合体のX線結晶構造と一致したことから,データの妥当性が示された.
合計266個の架橋から得られた情報を電子顕微鏡により得られた密度に反映するため,自動帰属のプログラムを開発した.このプログラムにより,まず,分子内架橋にもとづき各サブユニットを1~3個の剛体,たとえば,Rad3のような球状タンパク質は1個の剛体,Ssl2は2個の剛体に分割した.つぎに,G-ローブを電子顕微鏡により得られた密度の分布にもとづき12個の剛体に分割した.この12個の剛体に対して12の階乗とおりの帰属がありうるが,これら12の階乗個のモデルおのおのに対し架橋のパターンとの適合を評価した.最終的に得られたモデルは266個の架橋のうち263個の架橋と一致し,一致しなかった3個の架橋は自由度の高いフレキシブルなループにあった.自動帰属の結果は電子顕微鏡により得られた密度に対するドッキングと完全に一致し(図1b),また,ほかのグループから得られていたDNA-タンパク質結合マッピング11) など,生化学的な実験の結果を非常によく説明できるものであった.
4.転写開始の分子機構
この研究により,基本転写因子とRNAポリメラーゼIIがプロモーター領域のDNAに対しどのように結合するかがはじめて明らかにされた.TFIIHのヘリカーゼサブユニットであるSsl2はTATAボックスから30Å以上も離れて位置しており,TATAボックスとSsl2のあいだのDNA領域はタンパク質との結合により安定化されず溶媒に露出していて,そのため,電子顕微鏡によるマップでは円筒状の密度として観察された.Ssl2のATPase活性による負のトルク力によりこの円筒状の密度の2本鎖DNAはほどかれ,鋳型となるDNA鎖はすぐそばにあるRNAポリメラーゼIIのDNA結合クレフトに結合できると考えられた.
プロモーター領域のDNAはRNAポリメラーゼIIのDNA結合クレフトのちょうど真上に位置していたが,RNAポリメラーゼIIとは直接には結合しておらずG-ローブに強く結合していた(図2).RNAポリメラーゼIIのDNA結合クレフトの内部にはSsl2のC末端領域が結合しており,DNAとの結合を阻害する機構が観察された.RNAポリメラーゼIIのDNA結合クレフトにはDNAがちょうど結合する空間しかないことから,RNAポリメラーゼIIが鋳型となるDNA鎖に結合したのち,Ssl2のC末端領域はRNAポリメラーゼIIのDNA結合クレフトから排除されると考えられた(図2).そののち,転写が開始され,転写開始複合体はいくつもの構造の変化を経由して転写伸長のステージに入るが,これらの構造解析は現在進行中である.
おわりに
転写開始には基本転写因子とRNAポリメラーゼIIとが必要であることは20年以上もまえから知られていたが,それらが安定な転写開始複合体を形成していることは証明されていなかった.また,これまでのin vitroでの研究における転写活性は0.1~1.0%と本当にごくわずかで,放射能により標識したヌクレオチドを用いてようやく検知できるほどのものであった.筆者らの一連の研究においていちばん重要だったのは,転写開始複合体がゲルろ過カラムなどによる精製においても安定な複合体を保ち,クライオ電子顕微鏡やX線結晶回折により解析できるほどの安定性をもつことを証明して,高い転写活性をもつin vitro再構成系を確立したことである5,6).もちろん,転写開始複合体の構造解析も非常に困難であり,電子顕微鏡により得られた画像の処理法や化学架橋-質量分析法の技術改良は必須であった.
このようにして真核生物における転写開始はようやくin vitro再構成系が確立され,おそらく,つぎの10年から15年で転写の構造生物学はピークにいたると思われる.リボソームの構造が電子顕微鏡により30Åの分解能で解かれたのち,わずか10年たらずで原子構造までが解かれたことを考えれば,同じように転写開始複合体も原子構造が解明されることが期待できる.これらの研究はもちろん重要であるが,転写を研究する意義は“転写制御”の理解であることを忘れてはならないと思う.幸い,転写制御の分子機構の解明はまだ多くの地道な生化学的な研究が必要で,とくにin vitro再構成系においてはほとんどなにもされていない.1980年代から2000年代にかけて数人の研究者が転写の研究の土台を築いたように,今度は筆者の世代の研究者が転写制御の分野を切り開く必要があり,この分野はまだはじまったばかりである.
文 献
- Conaway, R. C. & Conaway, J. W.: General initiation factors for RNA polymerase II. Annu. Rev. Biochem., 62, 161-190 (1993)[PubMed]
- Roeder, R. G.: The role of general initiation factors in transcription by RNA polymerase II. Trends. Biochem. Sci., 21, 327-335 (1996)[PubMed]
- Kornberg, R. D.: The molecular basis of eukaryotic transcription. Proc. Natl. Acad. Sci. USA, 104, 12955-12961 (2007)[PubMed]
- Cramer, P., Bushnell, D. A., Fu, J. et al.: Architecture of RNA polymerase II and implications for the transcription mechanism. Science, 288, 640-649 (2000)[PubMed]
- Murakami, K., Calero, G., Brown, C. R. et al.: Formation and fate of a complete 31-protein RNA polymerase II transcription preinitiation complex. J. Biol. Chem., 288, 6325-6332 (2013)[PubMed]
- Murakami, K., Gibbons, B. J., Davis, R. E. et al.: Tfb6, a previously unidentified subunit of the general transcription factor TFIIH, facilitates dissociation of Ssl2 helicase after transcription initiation. Proc. Natl. Acad. Sci. USA, 109, 4816-4821 (2012)[PubMed]
- Gibbons, B. J., Brignole, E. J., Azubel, M. et al.: Subunit architecture of general transcription factor TFIIH. Proc. Natl. Acad. Sci. USA, 109, 1949-1954 (2012)[PubMed]
- Kostrewa, D., Zeller, M. E., Armache, K. J. et al.: RNA polymerase II-TFIIB structure and mechanism of transcription initiation. Nature, 462, 323-330 (2009)[PubMed]
- Liu, X., Bushnell, D. A., Wang, D. et al.: Structure of an RNA polymerase II-TFIIB complex and the transcription initiation mechanism. Science, 327, 206-209 (2010)[PubMed]
- Kalisman, N., Adams, C. M. & Levitt, M.: Subunit order of eukaryotic TRiC/CCT chaperonin by cross-linking, mass spectrometry, and combinatorial homology modeling. Proc. Natl. Acad. Sci. USA, 109, 2884-2889 (2012)[PubMed]
- Miller, G. & Hahn, S.: A DNA-tethered cleavage probe reveals the path for promoter DNA in the yeast preinitiation complex. Nat. Struct. Mol. Biol., 13, 603-610 (2006)[PubMed]
著者プロフィール
略歴:2004年 東京大学大学院理学系研究科 修了,2008年より米国Stanford大学 研究員.
研究テーマ:真核生物における転写制御.
© 2013 村上 健次 Licensed under CC 表示 2.1 日本
