アポトーシスを起こした細胞におけるXkr8によるホスファチジルセリンの露出
鈴木 淳・長田重一
(京都大学大学院医学研究科 医化学分野)
email:鈴木 淳,長田重一
DOI: 10.7875/first.author.2013.095
Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells.
Jun Suzuki, Daniel P. Denning, Eiichi Imanishi, H. Robert Horvitz, Shigekazu Nagata
Science, 341, 403-406 (2013)
アポトーシスを起こした細胞は,通常は細胞膜の内側に存在するリン脂質,ホスファチジルセリンを細胞の表面に露出し,自分は死んでいるという目印をつけることにより,マクロファージによって認識されて貪食される.しかしながら,このホスファチジルセリンの露出はどのような分子機構により担われているのか,まったくわかっていなかった.筆者らは,発現クローニングによりXkr8をアポトーシスのときにホスファチジルセリンの露出を実行するタンパク質として同定した.Xkr8をノックアウトしたマウスより調製した細胞,ならびに,Xkr8の発現がDNAメチル化により抑制されている白血病細胞はともに,アポトーシスの刺激のときにホスファチジルセリンを露出せず,マクロファージにより貪食されなかった.また,Xkr8はC末端にある細胞内領域がカスパーゼ3により切断されることで活性化し,通常は前駆体として存在することもわかった.さらに,Xkr8のホモログである線虫のCED-8にも同様の機能のあることが示されたことから,Xkr8/CED-8はホスファチジルセリンの露出を促進する,進化的に保存されたタンパク質であると結論した.
真核生物において細胞膜は外膜と内膜の2つの層から成り立っているが,細胞膜を構成するリン脂質は外膜と内膜とのあいだで非対称的に位置している.すなわち,ホスファチジルセリンは細胞膜の内側に,また,ホスファチジルコリンはおもに細胞膜の外側に位置している.ホスファチジルセリンの細胞膜の内側への輸送にはフリッパーゼ(aminophospholipid translocase)とよばれる酵素がかかわっており,ATPのエネルギーを用いてリン脂質の非対称性を維持している1).一方,血小板の活性化のときやアポトーシスのときにはこの非対称性が破綻しホスファチジルセリンが細胞の表面に露出する.このホスファチジルセリンの露出はフリッパーゼの不活性化のみでは不十分で,リン脂質を区別なく双方向に輸送するスクランブラーゼの活性が必要であると考えられていたが,その分子的な実体はまったく不明であった2).そこで,筆者らは,アポトーシスのときのリン脂質の双方向への輸送を担うタンパク質を同定することを目的として研究を進めた.
筆者らは,細胞外にCa2+のない状況においてCa2+イオノフォアA23187を用いると,生きた細胞が一過的にホスファチジルセリンを露出することをみつけた.そこで,Ba/F3細胞を1μMのA23187により刺激したのち,ホスファチジルセリンを高いレベルで露出した細胞を蛍光セルソーター(FACS)により分取した.そののち,A23187の濃度を段階的(500 nM,250 nM,125 nM)に下げながらCa2+イオノフォアによる刺激とFACSによる選別とをくり返し,ホスファチジルセリンを強く露出する細胞を濃縮した.最終的には,FACSによる細胞の分取を19回くり返すことにより,親細胞(PS0細胞)ではホスファチジルセリンをほとんど露出しない125 nMのA23187という条件においてもすみやかにホスファチジルセリンを露出する細胞(PS19細胞)を樹立することができた.そして,リン脂質を動かすタンパク質はおそらく高分子量だろうと考え,PS19細胞より2500 bp以上のサイズをもつcDNAを調製してcDNAライブラリーを作製し,Ba/F3細胞を宿主とした発現クローニングを行った.その結果,1つのアミノ酸残基の置換により活性の上昇した8回膜貫通タンパク質TMEM16Fを同定することができた.TMEM16FはCa2+の刺激ののちのリン脂質の双方向への輸送に関与していること,また,その生理的な役割としては,活性化した血小板においてホスファチジルセリンの露出に関与していることを見い出した3)(新着論文レビュー でも掲載).実際に,TMEM16Fノックアウトマウスに由来する胎仔胸腺細胞を不死化したのちCa2+で刺激しても,リン脂質の双方向への輸送はまったく起こらない.また,活性化した血小板においてホスファチジルセリンを露出できないことにより起こるScott症候群とよばれる血友病の患者では,TMEM16F遺伝子に変異が生じていた.
しかしながら,アポトーシスの刺激においてはTMEM16F欠損細胞も野生型の細胞と同様にホスファチジルセリンを露出したことから,アポトーシスのときには別のタンパク質が関与していると考えられた4).最初,TMEM16Fの属するTMEM16ファミリー(10つのメンバーより構成される)にアポトーシスのときにかかわるタンパク質があると考えた.そこで,Ca2+の刺激によりリン脂質の双方向の輸送が起こらなくなったTMEM16F欠損細胞にTMEM16ファミリーのメンバーを順に発現させてみたが,アポトーシスのときのホスファチジルセリンの露出に寄与していると考えられるメンバーはなかった4).このことより,アポトーシスのときにはまったくべつのタンパク質がはたらくと考え,再度,発現クローニングを行った.
筆者らはこれまでに,T細胞株であるWR19L細胞を用いてアポトーシスのときのホスファチジルセリンの露出がCa2+に依存することを確認していた.しかしながら,Ba/F3細胞から選別をくり返すことにより得たPS19細胞に由来する2500 bp以上のcDNAライブラリーを用いCa2+の刺激に応答するものとして同定された8つのcDNAは,すべてアポトーシスのときのホスファチジルセリンの露出にはかかわっていないTMEM16Fをコードするものであった.それでは,どのようにすればアポトーシスのときのホスファチジルセリンの露出にかかわるタンパク質を同定できるのだろうか? ここで,1000~2500 bpのcDNAにアポトーシスのときにかかわるタンパク質の遺伝子が含まれている可能性を考えた.アポトーシスのときにかかわるタンパク質はカスパーゼの刺激なども必要とするのではないかという不安もあったが,1000~2500 bpのサイズをもつcDNAを用いて作製したcDNAライブラリーをBa/F3細胞に発現させ,Ca2+の刺激ののちホスファチジルセリンを強く露出する細胞をFACSにより分取した.すると,5回の分取ののち,なんの刺激をしなくともホスファチジルセリンを露出する細胞を得た.この細胞のなかには,6回膜貫通領域をもつ機能未知のXkr8とよばれるタンパク質をコードするcDNAが組み込まれていた.TMEM16Fを同定したときとは異なり,Xkr8には変異は挿入されておらず,実際に,Ba/F3細胞にXkr8を過剰に発現させたところ,Ca2+の刺激がなくともホスファチジルセリンが露出した.現在までに,6種類の細胞にXkr8を過剰発現させてみたが,過剰発現によりホスファチジルセリンを構成的に露出するのはBa/F3細胞のみであった.つまり,発現クローニングにおいて宿主細胞としてBa/F3細胞を用いなければこのアプローチによりXkr8を同定できなかったことになり,幸運が味方してくれたと考えている.
Xkr8をWR19L細胞に過剰に発現させると,構成的にホスファチジルセリンを露出することはなかったが,アポトーシスの刺激によるホスファチジルセリンの露出が亢進した.一方で,Xkr8をノックダウンするとホスファチジルセリンの露出は抑制された.このとき,アポトーシスのときに活性化されるカスパーゼ3の活性化には影響がなかった.また,Xkr8をGFPと融合させて細胞に発現させると細胞膜に局在していた.以上より,Xkr8はホスファチジルセリンの露出の過程において,より直接的に関与していると考えられた.アポトーシスの刺激によりホスファチジルセリンを露出できない2つの白血病細胞株として,PLB985細胞5) およびRaji細胞6) が知られている.これらの細胞はXkr8をほとんど発現していなかった.そこで,これらの細胞にXkr8を外から導入すると,アポトーシスの刺激のときのホスファチジルセリンの露出は回復した.また,通常は細胞膜の内側に存在するホスファチジルエタノールアミンの露出,通常は細胞膜の外側に存在するホスファチジルコリンおよびスフィンゴミエリンの細胞内への取り込みも,Xkr8を導入することにより回復した.以上より,Xkr8がアポトーシスのときのリン脂質の双方向への輸送に関与していると結論した.このとき,アポトーシスのほかの指標であるDNAの断片化,ミトコンドリアに存在する酵素の不活性化,細胞の収縮には影響がなかったことから,Xkr8はアポトーシスのシグナル伝達においてその下流でホスファチジルセリンの露出に特異的にはたらいていると結論づけた.Xkr8を発現していないPLB-985細胞はアポトーシスにより死んでもマクロファージにほとんど貪食されないが,Xkr8を導入することでホスファチジルセリンを露出できるようになった死細胞は効率よく貪食された.これにより,Xkr8により露出したホスファチジルセリンが“eat-me-signal”として機能することが確認できた.
それでは,Xkr8はどのように活性化するのだろうか? Ca2+結合領域をコンピューターにより探索してもみつけることはできなかったが,C末端の細胞内領域にカスパーゼ3により認識される部位を見い出すことができた.実際に,アポトーシスの刺激をくわえるとXkr8はこの部位において切断され,この部位にカスパーゼ3が認識できなくなる変異を導入するとXkr8は切断されなくなり,ホスファチジルセリンを露出させる能力も失われた.つまり,Xkr8は正常な細胞では前駆体として存在し,アポトーシスのときにカスパーゼにより切断されて活性化されると結論づけた.
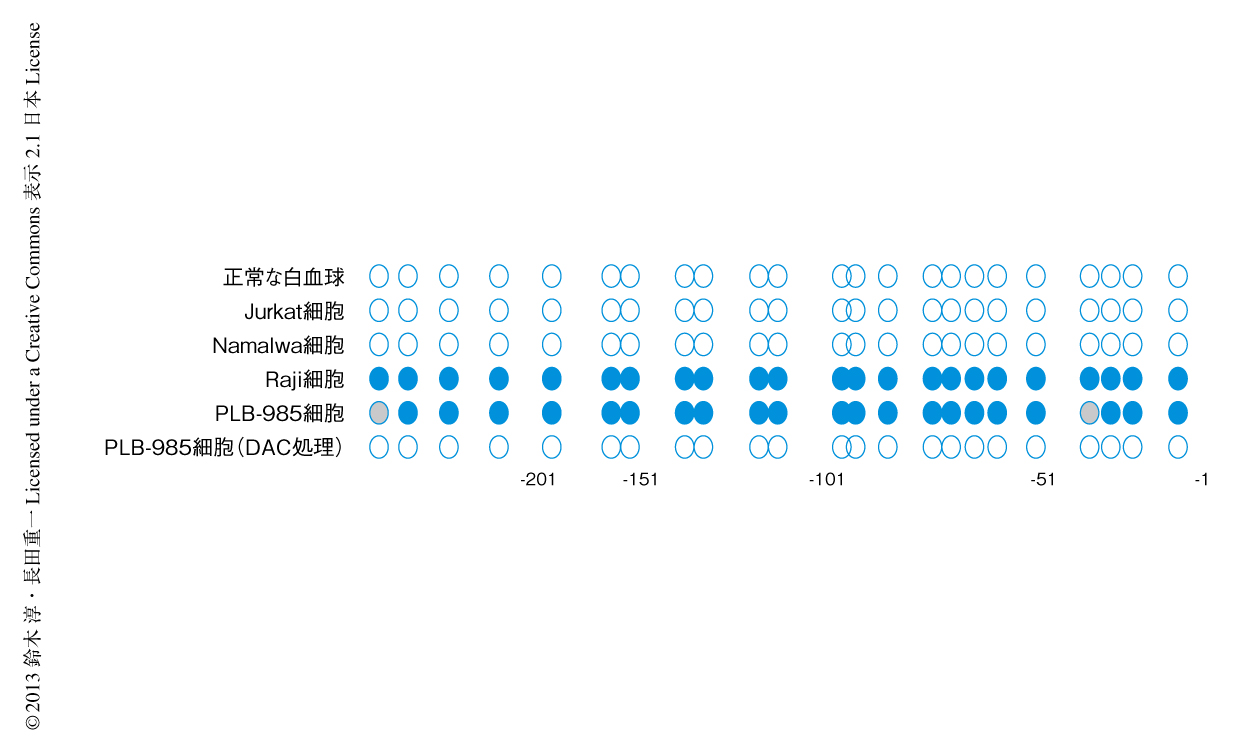
PLB-985細胞およびRaji細胞はともにXkr8をほとんど発現していないが,これはなぜなのだろうか? Xkr8遺伝子のゲノム領域(約10 kbp)を遺伝子クローニングして解析したが,大きな欠失や挿入はみあたらなかった.一方,コンピューターによる検索により,Xkr8遺伝子のプロモーター領域は非常にCGに富み,CpCアイランドを形成していることが判明した.そこで,このプロモーター領域のシトシンはDNAメチル化されているのではないかと考え,bisulfite法(通常のシトシンはbisulfiteによりウラシルに変換されるが,メチル化されているシトシンは変換されないことを原理とする)を用いて調べたところ,高頻度でDNAメチル化されていた(図1).一方で,ほかの白血病細胞であるJurkat細胞やNamalwa細胞,また,通常の白血球ではXkr8遺伝子のプロモーター領域はDNAメチル化されていなかった.PLB-985細胞をDNAメチルトランスフェラーゼの阻害剤である5-アザ-2’-デオキシシチジンにより7日間にわたり処理するとDNAメチル化は消失し,Xkr8のmRNAレベルの発現も上昇した.また,このときアポトーシスの刺激をするとホスファチジルセリンの露出が回復した.このように,ある種の白血病細胞においてはXkr8遺伝子のプロモーター領域がDNAメチル化されることによりその発現が抑制され,ホスファチジルセリンが露出されないことがわかった.
Xkr8の線虫のホモログであるCED-8は細胞死のタイミングを制御するタンパク質として結論づけられていたが,その詳細な機能についてはわかっていなかった7).死細胞の貪食に関与するCED-1やCED-5の変異体では,貪食されなかった死細胞が組織から剥離した細胞として検出される8).CED-8の変異体においても組織から剥離した細胞が検出され,その数はCED-1やCED-5の変異体と掛け合わせることにより増加した.そして,CED-1やCED-5の変異により生じた組織から剥離した細胞はホスファチジルセリンを露出していたが,CED-8に変異を導入することによりホスファチジルセリンの露出はみられなくなった.以上より,線虫のced変異体の責任タンパク質のなかでその機能が長年にわたり不明であったCED-8は,ホスファチジルセリンの露出を制御していることが明らかになった.
この研究により,長年の疑問であったアポトーシスのときのホスファチジルセリンの露出にかかわるタンパク質としてXkr8が同定された.Xkr8はヒトにおいては9つのメンバー,マウスにおいては8つのメンバーにより構成されるXkrファミリーに属しており,今後,ほかのXkrファミリーのメンバーにおいても同様の活性があるのか調べる必要があるだろう.また,アポトーシスのときのホスファチジルセリンの露出にはCa2+が必要であると考えられているが9),筆者らの実験結果では,WR19L細胞においてはCa2+に依存性であるが,Ba/F3細胞においてはCa2+を要求しなかった.Ba/F3細胞においてはXkr8を過剰に発現するだけでホスファチジルセリンが露出したことから,Ca2+により制御される未同定のXkr8結合タンパク質が欠損しているのかもしれないと考えている.最後に,Ca2+の刺激により活性化されるTMEM16F,および,アポトーシスの刺激により活性化されるXkr8が,直接的にリン脂質を動かしているかどうかを,試験管内の再構成系などによりはっきりさせる必要があるだろう.
略歴:2007年 大阪大学大学院医学系研究科博士課程 修了,同年 京都大学大学院医学研究科 博士研究員を経て,2010年より同 助教.
研究テーマ:細胞膜におけるリン脂質ホスファチジルセリンの露出の機構.
関心事:細胞膜におけるリン脂質の制御機構,また,その生理的な役割.
長田 重一(Shigekazu Nagata)
京都大学大学院医学研究科 教授.
研究室URL:http://www2.mfour.med.kyoto-u.ac.jp/~nagata/index.html
© 2013 鈴木 淳・長田重一 Licensed under CC 表示 2.1 日本
(京都大学大学院医学研究科 医化学分野)
email:鈴木 淳,長田重一
DOI: 10.7875/first.author.2013.095
Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells.
Jun Suzuki, Daniel P. Denning, Eiichi Imanishi, H. Robert Horvitz, Shigekazu Nagata
Science, 341, 403-406 (2013)
要 約
アポトーシスを起こした細胞は,通常は細胞膜の内側に存在するリン脂質,ホスファチジルセリンを細胞の表面に露出し,自分は死んでいるという目印をつけることにより,マクロファージによって認識されて貪食される.しかしながら,このホスファチジルセリンの露出はどのような分子機構により担われているのか,まったくわかっていなかった.筆者らは,発現クローニングによりXkr8をアポトーシスのときにホスファチジルセリンの露出を実行するタンパク質として同定した.Xkr8をノックアウトしたマウスより調製した細胞,ならびに,Xkr8の発現がDNAメチル化により抑制されている白血病細胞はともに,アポトーシスの刺激のときにホスファチジルセリンを露出せず,マクロファージにより貪食されなかった.また,Xkr8はC末端にある細胞内領域がカスパーゼ3により切断されることで活性化し,通常は前駆体として存在することもわかった.さらに,Xkr8のホモログである線虫のCED-8にも同様の機能のあることが示されたことから,Xkr8/CED-8はホスファチジルセリンの露出を促進する,進化的に保存されたタンパク質であると結論した.
はじめに
真核生物において細胞膜は外膜と内膜の2つの層から成り立っているが,細胞膜を構成するリン脂質は外膜と内膜とのあいだで非対称的に位置している.すなわち,ホスファチジルセリンは細胞膜の内側に,また,ホスファチジルコリンはおもに細胞膜の外側に位置している.ホスファチジルセリンの細胞膜の内側への輸送にはフリッパーゼ(aminophospholipid translocase)とよばれる酵素がかかわっており,ATPのエネルギーを用いてリン脂質の非対称性を維持している1).一方,血小板の活性化のときやアポトーシスのときにはこの非対称性が破綻しホスファチジルセリンが細胞の表面に露出する.このホスファチジルセリンの露出はフリッパーゼの不活性化のみでは不十分で,リン脂質を区別なく双方向に輸送するスクランブラーゼの活性が必要であると考えられていたが,その分子的な実体はまったく不明であった2).そこで,筆者らは,アポトーシスのときのリン脂質の双方向への輸送を担うタンパク質を同定することを目的として研究を進めた.
1.TMEM16Fの同定
筆者らは,細胞外にCa2+のない状況においてCa2+イオノフォアA23187を用いると,生きた細胞が一過的にホスファチジルセリンを露出することをみつけた.そこで,Ba/F3細胞を1μMのA23187により刺激したのち,ホスファチジルセリンを高いレベルで露出した細胞を蛍光セルソーター(FACS)により分取した.そののち,A23187の濃度を段階的(500 nM,250 nM,125 nM)に下げながらCa2+イオノフォアによる刺激とFACSによる選別とをくり返し,ホスファチジルセリンを強く露出する細胞を濃縮した.最終的には,FACSによる細胞の分取を19回くり返すことにより,親細胞(PS0細胞)ではホスファチジルセリンをほとんど露出しない125 nMのA23187という条件においてもすみやかにホスファチジルセリンを露出する細胞(PS19細胞)を樹立することができた.そして,リン脂質を動かすタンパク質はおそらく高分子量だろうと考え,PS19細胞より2500 bp以上のサイズをもつcDNAを調製してcDNAライブラリーを作製し,Ba/F3細胞を宿主とした発現クローニングを行った.その結果,1つのアミノ酸残基の置換により活性の上昇した8回膜貫通タンパク質TMEM16Fを同定することができた.TMEM16FはCa2+の刺激ののちのリン脂質の双方向への輸送に関与していること,また,その生理的な役割としては,活性化した血小板においてホスファチジルセリンの露出に関与していることを見い出した3)(新着論文レビュー でも掲載).実際に,TMEM16Fノックアウトマウスに由来する胎仔胸腺細胞を不死化したのちCa2+で刺激しても,リン脂質の双方向への輸送はまったく起こらない.また,活性化した血小板においてホスファチジルセリンを露出できないことにより起こるScott症候群とよばれる血友病の患者では,TMEM16F遺伝子に変異が生じていた.
しかしながら,アポトーシスの刺激においてはTMEM16F欠損細胞も野生型の細胞と同様にホスファチジルセリンを露出したことから,アポトーシスのときには別のタンパク質が関与していると考えられた4).最初,TMEM16Fの属するTMEM16ファミリー(10つのメンバーより構成される)にアポトーシスのときにかかわるタンパク質があると考えた.そこで,Ca2+の刺激によりリン脂質の双方向の輸送が起こらなくなったTMEM16F欠損細胞にTMEM16ファミリーのメンバーを順に発現させてみたが,アポトーシスのときのホスファチジルセリンの露出に寄与していると考えられるメンバーはなかった4).このことより,アポトーシスのときにはまったくべつのタンパク質がはたらくと考え,再度,発現クローニングを行った.
2.Xkr8の同定
筆者らはこれまでに,T細胞株であるWR19L細胞を用いてアポトーシスのときのホスファチジルセリンの露出がCa2+に依存することを確認していた.しかしながら,Ba/F3細胞から選別をくり返すことにより得たPS19細胞に由来する2500 bp以上のcDNAライブラリーを用いCa2+の刺激に応答するものとして同定された8つのcDNAは,すべてアポトーシスのときのホスファチジルセリンの露出にはかかわっていないTMEM16Fをコードするものであった.それでは,どのようにすればアポトーシスのときのホスファチジルセリンの露出にかかわるタンパク質を同定できるのだろうか? ここで,1000~2500 bpのcDNAにアポトーシスのときにかかわるタンパク質の遺伝子が含まれている可能性を考えた.アポトーシスのときにかかわるタンパク質はカスパーゼの刺激なども必要とするのではないかという不安もあったが,1000~2500 bpのサイズをもつcDNAを用いて作製したcDNAライブラリーをBa/F3細胞に発現させ,Ca2+の刺激ののちホスファチジルセリンを強く露出する細胞をFACSにより分取した.すると,5回の分取ののち,なんの刺激をしなくともホスファチジルセリンを露出する細胞を得た.この細胞のなかには,6回膜貫通領域をもつ機能未知のXkr8とよばれるタンパク質をコードするcDNAが組み込まれていた.TMEM16Fを同定したときとは異なり,Xkr8には変異は挿入されておらず,実際に,Ba/F3細胞にXkr8を過剰に発現させたところ,Ca2+の刺激がなくともホスファチジルセリンが露出した.現在までに,6種類の細胞にXkr8を過剰発現させてみたが,過剰発現によりホスファチジルセリンを構成的に露出するのはBa/F3細胞のみであった.つまり,発現クローニングにおいて宿主細胞としてBa/F3細胞を用いなければこのアプローチによりXkr8を同定できなかったことになり,幸運が味方してくれたと考えている.
3.Xkr8によるホスファチジルセリンの露出
Xkr8をWR19L細胞に過剰に発現させると,構成的にホスファチジルセリンを露出することはなかったが,アポトーシスの刺激によるホスファチジルセリンの露出が亢進した.一方で,Xkr8をノックダウンするとホスファチジルセリンの露出は抑制された.このとき,アポトーシスのときに活性化されるカスパーゼ3の活性化には影響がなかった.また,Xkr8をGFPと融合させて細胞に発現させると細胞膜に局在していた.以上より,Xkr8はホスファチジルセリンの露出の過程において,より直接的に関与していると考えられた.アポトーシスの刺激によりホスファチジルセリンを露出できない2つの白血病細胞株として,PLB985細胞5) およびRaji細胞6) が知られている.これらの細胞はXkr8をほとんど発現していなかった.そこで,これらの細胞にXkr8を外から導入すると,アポトーシスの刺激のときのホスファチジルセリンの露出は回復した.また,通常は細胞膜の内側に存在するホスファチジルエタノールアミンの露出,通常は細胞膜の外側に存在するホスファチジルコリンおよびスフィンゴミエリンの細胞内への取り込みも,Xkr8を導入することにより回復した.以上より,Xkr8がアポトーシスのときのリン脂質の双方向への輸送に関与していると結論した.このとき,アポトーシスのほかの指標であるDNAの断片化,ミトコンドリアに存在する酵素の不活性化,細胞の収縮には影響がなかったことから,Xkr8はアポトーシスのシグナル伝達においてその下流でホスファチジルセリンの露出に特異的にはたらいていると結論づけた.Xkr8を発現していないPLB-985細胞はアポトーシスにより死んでもマクロファージにほとんど貪食されないが,Xkr8を導入することでホスファチジルセリンを露出できるようになった死細胞は効率よく貪食された.これにより,Xkr8により露出したホスファチジルセリンが“eat-me-signal”として機能することが確認できた.
それでは,Xkr8はどのように活性化するのだろうか? Ca2+結合領域をコンピューターにより探索してもみつけることはできなかったが,C末端の細胞内領域にカスパーゼ3により認識される部位を見い出すことができた.実際に,アポトーシスの刺激をくわえるとXkr8はこの部位において切断され,この部位にカスパーゼ3が認識できなくなる変異を導入するとXkr8は切断されなくなり,ホスファチジルセリンを露出させる能力も失われた.つまり,Xkr8は正常な細胞では前駆体として存在し,アポトーシスのときにカスパーゼにより切断されて活性化されると結論づけた.
4.Xkr8遺伝子の発現抑制
PLB-985細胞およびRaji細胞はともにXkr8をほとんど発現していないが,これはなぜなのだろうか? Xkr8遺伝子のゲノム領域(約10 kbp)を遺伝子クローニングして解析したが,大きな欠失や挿入はみあたらなかった.一方,コンピューターによる検索により,Xkr8遺伝子のプロモーター領域は非常にCGに富み,CpCアイランドを形成していることが判明した.そこで,このプロモーター領域のシトシンはDNAメチル化されているのではないかと考え,bisulfite法(通常のシトシンはbisulfiteによりウラシルに変換されるが,メチル化されているシトシンは変換されないことを原理とする)を用いて調べたところ,高頻度でDNAメチル化されていた(図1).一方で,ほかの白血病細胞であるJurkat細胞やNamalwa細胞,また,通常の白血球ではXkr8遺伝子のプロモーター領域はDNAメチル化されていなかった.PLB-985細胞をDNAメチルトランスフェラーゼの阻害剤である5-アザ-2’-デオキシシチジンにより7日間にわたり処理するとDNAメチル化は消失し,Xkr8のmRNAレベルの発現も上昇した.また,このときアポトーシスの刺激をするとホスファチジルセリンの露出が回復した.このように,ある種の白血病細胞においてはXkr8遺伝子のプロモーター領域がDNAメチル化されることによりその発現が抑制され,ホスファチジルセリンが露出されないことがわかった.
5.Xkr8のホモログCED-8
Xkr8の線虫のホモログであるCED-8は細胞死のタイミングを制御するタンパク質として結論づけられていたが,その詳細な機能についてはわかっていなかった7).死細胞の貪食に関与するCED-1やCED-5の変異体では,貪食されなかった死細胞が組織から剥離した細胞として検出される8).CED-8の変異体においても組織から剥離した細胞が検出され,その数はCED-1やCED-5の変異体と掛け合わせることにより増加した.そして,CED-1やCED-5の変異により生じた組織から剥離した細胞はホスファチジルセリンを露出していたが,CED-8に変異を導入することによりホスファチジルセリンの露出はみられなくなった.以上より,線虫のced変異体の責任タンパク質のなかでその機能が長年にわたり不明であったCED-8は,ホスファチジルセリンの露出を制御していることが明らかになった.
おわりに
この研究により,長年の疑問であったアポトーシスのときのホスファチジルセリンの露出にかかわるタンパク質としてXkr8が同定された.Xkr8はヒトにおいては9つのメンバー,マウスにおいては8つのメンバーにより構成されるXkrファミリーに属しており,今後,ほかのXkrファミリーのメンバーにおいても同様の活性があるのか調べる必要があるだろう.また,アポトーシスのときのホスファチジルセリンの露出にはCa2+が必要であると考えられているが9),筆者らの実験結果では,WR19L細胞においてはCa2+に依存性であるが,Ba/F3細胞においてはCa2+を要求しなかった.Ba/F3細胞においてはXkr8を過剰に発現するだけでホスファチジルセリンが露出したことから,Ca2+により制御される未同定のXkr8結合タンパク質が欠損しているのかもしれないと考えている.最後に,Ca2+の刺激により活性化されるTMEM16F,および,アポトーシスの刺激により活性化されるXkr8が,直接的にリン脂質を動かしているかどうかを,試験管内の再構成系などによりはっきりさせる必要があるだろう.
文 献
- Daleke, D.: Phospholipid flippases. J. Biol. Chem., 282, 821-825 (2007)[PubMed]
- Balasubramanian, K. & Schroit, A. J.: Aminophospholipid asymmetry: a matter of life and death. Annu. Rev. Physiol., 65, 701-734 (2003)[PubMed]
- Suzuki, J., Umeda, M., Sims, P. J. et al.: Calcium-dependent phospholipid scrambling by TMEM16F. Nature, 468, 834-838 (2010)[PubMed] [新着論文レビュー]
- Suzuki, J., Fujii, T., Imao, T. et al.: Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J. Biol. Chem., 288, 13305-11316 (2013)[PubMed]
- Fadok, V. A., de Cathelineau, A., Daleke, D. L. et al.: Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J. Biol. Chem., 276, 1071-1077 (2001)[PubMed]
- Fadeel, B., Gleiss, B., Hogstrand, K. et al.: Phosphatidylserine exposure during apoptosis is a cell-type-specific event and does not correlate with plasma membrane phospholipid scramblase expression. Biochem. Biophys. Res. Commun., 266, 504-511 (1999)[PubMed]
- Stanfield, G. M. & Horvitz, H. R.: The ced-8 gene controls the timing of programmed cell deaths in C. elegans. Mol. Cell, 5, 423-433 (2000)[PubMed]
- Denning, D. P., Hatch, V. & Horvitz, H. R.: Programmed elimination of cells by caspase-independent cell extrusion in C. elegans. Nature, 488, 226-230 (2012)[PubMed]
- Bevers, E. M. & Williamson, P. L.: Phospholipid scramblase: an update. FEBS Lett., 584, 2724-2730 (2010)[PubMed]
著者プロフィール
略歴:2007年 大阪大学大学院医学系研究科博士課程 修了,同年 京都大学大学院医学研究科 博士研究員を経て,2010年より同 助教.
研究テーマ:細胞膜におけるリン脂質ホスファチジルセリンの露出の機構.
関心事:細胞膜におけるリン脂質の制御機構,また,その生理的な役割.
長田 重一(Shigekazu Nagata)
京都大学大学院医学研究科 教授.
研究室URL:http://www2.mfour.med.kyoto-u.ac.jp/~nagata/index.html
© 2013 鈴木 淳・長田重一 Licensed under CC 表示 2.1 日本
