肝臓のグルコキナーゼは神経ネットワークを介し褐色脂肪組織における熱産生を制御することにより肥満になりやすさをつかさどる
突田壮平・山田哲也・片桐秀樹
(東北大学大学院医学系研究科 附属創生応用医学研究センター代謝疾患学分野)
email:突田壮平,山田哲也
DOI: 10.7875/first.author.2013.003
Hepatic glucokinase modulates obesity predisposition by regulating BAT thermogenesis via neural signals.
Sohei Tsukita, Tetsuya Yamada, Kenji Uno, Kei Takahashi, Keizo Kaneko, Yasushi Ishigaki, Junta Imai, Yutaka Hasegawa, Shojiro Sawada, Hisamitsu Ishihara, Yoshitomo Oka, Hideki Katagiri
Cell Metabolism, 16, 825-832 (2012)
体重は本質的には摂取エネルギーと消費エネルギーとの収支により決定されるが,日々の食事量や活動量の変動がそのまま直接的に体重の変化に反映されるわけではない.近年,脳をオーガナイザーとする多くの臓器間相互作用がフィーバック機構として機能し,過剰なエネルギーの摂取に対する体重の恒常性の維持に寄与していることが明らかになった.一方,飽食の現代において肥満人口が増加の一途をたどっていることから,エネルギーの摂取に対しフィードフォワード機構としてはたらき体重の増加に寄与する臓器間相互作用の存在が想定されるものの,その存在は明らかでなかった.この研究では,この一端を説明しうる,肝臓からの新たな臓器間神経ネットワークを見い出した.エネルギーの摂取過剰の条件において,グルコキナーゼの発現上昇にともない肝臓における糖代謝に変化が生じると,その変化により惹起されたシグナルが迷走神経の求心路を介し伝達される.これをうけ,延髄から褐色脂肪組織へといたる交感神経の活性が低下し,褐色脂肪組織における熱産生が抑制され脂肪の蓄積が促進される.この機構はエネルギーの摂取増加に呼応するエネルギー消費の低下をつかさどっており,その観点から個体レベルにおける倹約機構と考えられた.食事を満足に得ることのできなかった時代において生存に有利にはたらいたと考えられるこの機構は,皮肉にも,飽食の現代において肥満に舵を切る分子機構としてはたらいているものと想定された.さらに,この機構のはたらきの違いが,肥満のなりやすさに関するマウスの系統の違いを規定する一因となっていることが示された.
肥満は糖尿病,高脂血症,高血圧症など生活習慣病に共通する要因であり,その点からも肥満の研究,すなわち,体重の制御機構の解明は重要である.体重の恒常性を維持することの本質的な意義は,食物をつねに得ることができるかどうか不確かな環境において体にすぐに利用できるエネルギー源を蓄えておくことにあったと考えられる.一方で,脂肪の過剰な蓄積は個体の活動性を損ない捕食や外敵からの回避において不利益をもたらすことになる.したがって,エネルギー源の貯蓄量と補給量とにもとづき摂食行動およびエネルギー消費量を適正に制御する必要があり,視床下部はこの分子機構において中心的な役割を担っている.たとえば,蓄積する中性脂肪の量に応じ白色脂肪細胞から分泌されるレプチンは,視床下部に作用し摂食の抑制およびエネルギー消費の亢進をもたらすことによりエネルギーの摂取過剰に対するフィードバック機構を構成することが知られている1).また,レプチンのような液性因子にくわえ,近年,神経ネットワークもエネルギー代謝の恒常性の維持に重要な役割をはたしていることが明らかになってきた2).いずれにしても,エネルギー摂取の増加に対し体重の恒常性を維持するよう機能するフィードバック機構が厳格に機能するならば体重の増加は起こりえず,したがって,肥満は生じないはずである.しかしながら,肥満人口が増加の一途をたどっている状況を鑑みると,体重の恒常性の維持の機構は容易に破綻する,もしくは,体重の増加にむけ機能するフィードフォワード機構の存在が想起される.
筆者らは,後者の観点から,肝臓における糖代謝の変化が全身のエネルギー代謝に及ぼす影響を検討することにした.なぜなら,肝臓において特異的にグルコキナーゼを過剰発現するトランスジェニックマウスは肥満の表現型を示すことが報告されており3),肝臓における糖代謝が全身のエネルギー代謝に影響を及ぼすことが示唆されていたからである.グルコキナーゼはインスリンにより強力に発現誘導され,肝細胞に取り込まれたグルコースをグルコース6-リン酸に変換する反応を触媒する酵素である.この研究では,アデノウイルス遺伝子発現系を用いてマウスの肝臓において選択的にグルコキナーゼを過剰発現させることにより肝臓における解糖系を亢進し,肝臓における糖代謝の変化が全身のエネルギー収支,とくに,褐色脂肪組織におけるエネルギー消費に及ぼす影響を検討した.褐色脂肪組織は熱産生タンパク質であるUCP1を特異的に発現し,酸化的リン酸化を脱共役させることによりエネルギーを熱として消費するという機能をもつ.これまで,褐色脂肪組織はげっ歯類やヒトの新生児に特異的に存在するものとされていたが,最近,ヒトの成人においても機能的に活発な褐色脂肪組織の存在することや,褐色脂肪組織の量や活性が低いと肥満につながる可能性のあることが報告され,注目をあつめている4).
C57BL/6マウスに高脂肪かつ高スクロース食を負荷したところ,肝臓における内因性のグルコキナーゼの発現は負荷1週目より著明に上昇した.そこで,この負荷の早期より生じる肝臓におけるグルコキナーゼの発現上昇に着目し,個体レベルでのエネルギー代謝に及ぼす影響を調べることにした.通常食により飼育しているマウスの肝臓にアデノウイルスベクターを用いてグルコキナーゼを過剰発現させたところ,その発現量に依存して肝臓におけるグリコーゲンおよび中性脂肪の量が増加した.マイクロアレイを用いた遺伝子発現解析ではグリコーゲン合成および脂肪合成に関与する酵素の遺伝子の発現上昇が確認された.このマウスでは,対照となる野生型マウスと比較して,摂餌量および活動量に差は認められなかった.また,プローブを用いて直接的に測定した肝臓の深部の温度にも差は認められず,赤外線サーモグラフィー画像においても肝臓の表面の温度に差は認められなかった.これらのことより,グルコキナーゼの発現上昇は肝臓における熱産生には影響を及ぼさないことが判明した.
肝臓においてグルコキナーゼを過剰発現させたマウスにおいて,肩甲骨のあいだに存在する褐色脂肪組織の組織像を検討したところ,興味深いことに,脂肪滴の直径が増加しており熱産生の低下が示唆された.また,肝臓におけるグルコキナーゼの発現量に依存して,褐色脂肪組織における熱産生関連遺伝子UCP1遺伝子,PGC-1α遺伝子,D2遺伝子の発現が低下しており,これは脂肪滴の直径の増大と相応した.これら熱産生遺伝子は交感神経系の活性によりその発現が制御されていることが知られており,交感神経系の活性の低下が強く示唆された.実際に,褐色脂肪組織におけるノルアドレナリンの代謝回転を測定したところ低下しており,褐色脂肪組織にいたる交感神経の活性が低下していることが確認された.さらに,褐色脂肪組織において熱産生を制御する交感神経のプレモーターニューロンが存在する延髄の吻側の縫線核において5),神経活性化のマーカーであるc-fosはmRNAレベルおよびタンパク質レベルにおいて低下していた.これらのことより,肝臓におけるグルコキナーゼの発現上昇は,少なくとも,延髄のプレモーターニューロンのレベルにおいて交感神経の活性を下方制御し,褐色脂肪組織における熱産生を抑制しているものと考えられた.
褐色脂肪組織における熱産生の低下が個体レベルでのエネルギー消費に及ぼす影響を,ノルアドレナリンの投与に対する酸素消費量の増加量(適応性非ふるえ熱産生能6))により評価した.肝臓においてグルコキナーゼを過剰発現させたマウスではノルアドレナリンの投与に対する酸素消費量の増加反応が低下しており,個体レベルでのエネルギー消費の低下していることが確認された.さらに,このマウスを温度的中性条件である28℃において飼育すると,肝臓におけるグルコキナーゼの発現量に依存して体重および白色脂肪の重量は増加した.以上より,肝臓におけるグルコキナーゼの発現上昇により褐色脂肪組織における熱産生が低下し,個体レベルでのエネルギー消費が減少し体重および脂肪量の増加することが示された.
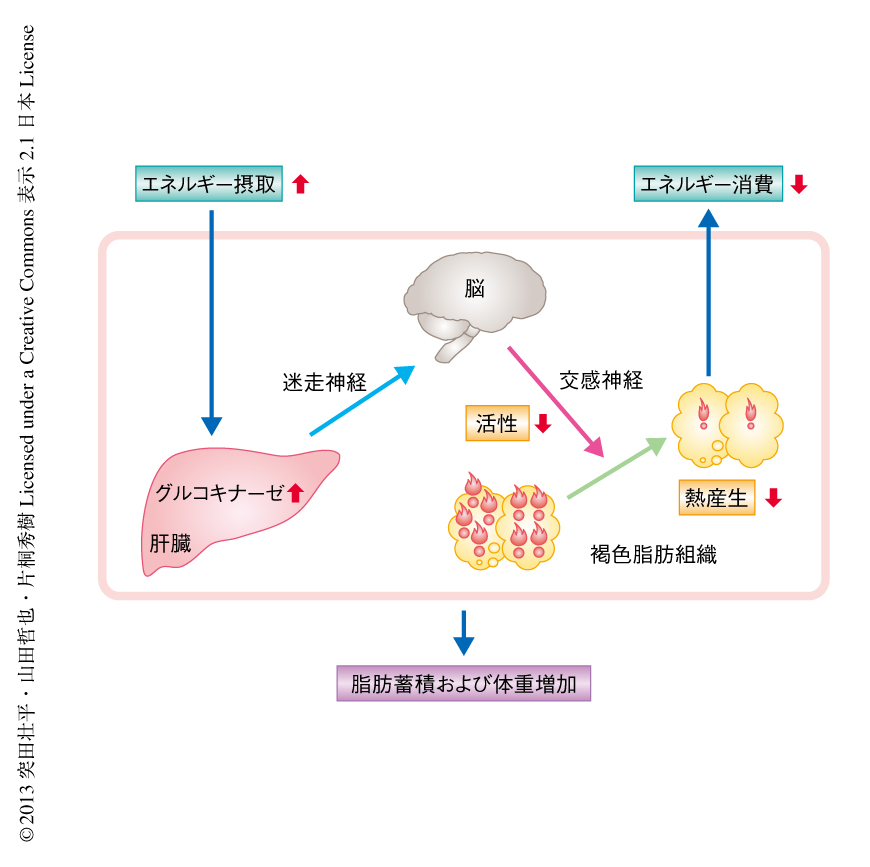
肝臓におけるグルコキナーゼの発現上昇は交感神経の活性を下方制御し褐色脂肪組織における熱産生を抑制するわけだが,シグナルはどのような経路を介し肝臓から脳へと伝達されるのだろうか? 肝臓における代謝のシグナルが迷走神経の求心路を介し中枢へと伝達されることが報告されていたことから7,8),肝臓と中枢とをつなぐ経路として迷走神経の求心路の関与を考えた.迷走神経の肝臓枝を選択的に切断したのち肝臓においてグルコキナーゼを過剰発現させたところ,迷走神経を切断しないマウスで観察された褐色脂肪組織における脂肪滴の直径の増大,熱産生関連遺伝子の発現低下,縫線核におけるc-fosの発現低下などの変化は認められなくなった.このことから,褐色脂肪組織における熱産生の抑制は肝臓の発する迷走神経のシグナルを介すものと考えられた(図1).
レプチンは視床下部に作用し摂食の抑制およびエネルギー消費の亢進をもたらすことにより,エネルギーの摂取過剰に対しフィードバック機構としてはたらき,体重の恒常性の維持に重要な役割を担うことが知られている.そこで,レプチンによるフィードバック機構と肝臓と褐色脂肪組織とのあいだのフィードフォワード機構との相互作用について検討した.肝臓においてグルコキナーゼを過剰発現させたマウスの腹腔にレプチンを投与したところ,対照となる野生型マウスと同等の摂食の抑制効果が認められた.さらに,この両方のマウスにおいて,レプチンの投与により視床下部の弓状核におけるc-fosの発現上昇,摂食関連ペプチドのうち摂食抑制作用をもつPOMCの発現上昇および摂食亢進作用をもつNPYの発現抑制が認められた.以上より,肝臓のグルコキナーゼにより誘導される迷走神経のシグナルは,レプチンによる摂食の抑制効果に影響をあたえないことが判明した.一方,レプチンの投与により野生型マウスにおいて認められた縫線核におけるc-fos発現上昇や褐色脂肪組織における熱産生関連遺伝子の発現上昇は,肝臓においてグルコキナーゼを過剰発現させたマウスにおいて認められなかった.したがって,この迷走神経のシグナルは,縫線核-交感神経-褐色脂肪組織とつながる熱産生経路を抑制するものと考えられた.さらに,縫線核の上流において褐色脂肪組織へといたる交感神経の活性の制御に関与する視床下部の室傍核9) に対する,レプチンの投与による活性化は,肝臓においてグルコキナーゼを過剰発現させたマウスにおいても影響をうけなったことから,このシグナルはレプチンによるエネルギー消費の亢進作用を視床下部より下流において抑制するものと考えられた(図2).

肝臓におけるグルコキナーゼの発現上昇に端を発する肝臓と褐色脂肪組織との組織間の機構は,病態生理的にはどのような役割をはたしているのだろうか? 過剰なエネルギーの摂取により肥満をきたしやすいC57BL/6マウスおよびAKRマウスと,肥満をきたしにくいSWR/JマウスおよびA/Jマウスに対し10),高脂肪かつ高スクロース食を1週間にわたり負荷し,肝臓における内因性のグルコキナーゼの発現および褐色脂肪組織におけるUCP1遺伝子の発現を比較した.高脂肪かつ高スクロース食の負荷により,肝臓におけるグルコキナーゼの発現は,肥満をきたしやすい系統のマウスにおいて顕著に上昇した.一方,褐色脂肪組織におけるUCP1遺伝子の発現は,肥満をきたしにくい系統のマウスにおいて上昇した.すなわち,肝臓におけるグルコキナーゼの発現と褐色脂肪組織におけるUCP1遺伝子の発現のあいだには逆相関が認められた.
肝臓と褐色脂肪組織との組織間の機構が肥満の指向性に関与している可能性を考え,以下の実験を行った.まず,肥満をきたしにくい系統であるSWR/Jマウスの肝臓においてグルコキナーゼを過剰発現させたところ,褐色脂肪組織における熱産生は低下し体重の増加が促進された.つぎに,肥満をきたしやすい系統であるC57BL/6マウスにおいて脂肪かつ高スクロース食の負荷により生じる肝臓における内因性のグルコキナーゼの発現上昇に対しこれをshRNAによりノックダウンしたところ,褐色脂肪組織における熱産生は増加し体重の増加は抑制された.以上より,高脂肪かつ高スクロース食の負荷にともなう肝臓における内因性のグルコキナーゼの発現上昇は,熱産生能を制御することにより肥満の指向性を決める一因となると考えられた.
この研究により,これまで見い出されていなかった,エネルギーの過剰な摂取に対する体重制御におけるフィードフォワード機構の存在が明らかになった.食物の供給が不安定な自然環境では,余分に摂取したエネルギーを脂肪として効率よく蓄積することは飢餓に直面した際に個体の生存に有利にはたらくと思われ,その観点から,この機構は個体レベルでの“倹約機構”と考えられる.一方,現代社会のような慢性的に過栄養な環境においてこの機構は体重増加のきっかけとなり,ひいては,肥満の発症に関与するものと考えられる.
今後の課題として,肝臓における糖代謝の変化がどのような分子機構を介し迷走神経の活動を変化させるのか,ヒトの成人においてもこの倹約機構が機能しているか,など明らかにすべき点は多い.このフィードフォワード機構に介入し,肥満者において低下している褐色脂肪組織における熱産生を活性化することができれば,過栄養時代における新たな肥満症の治療法や予防法の確立につながるものと考えられる.
略歴:2011年 東北大学大学院医学系研究科 修了,同年より同 助手.
研究テーマ:糖代謝およびエネルギー代謝の制御機構.
抱負:糖尿病や肥満症の病態の解明また治療法の開発に貢献したい.日常の診療では体重増加を助長しない血糖降下の手助けをしたい.
山田 哲也(Tetsuya Yamada)
東北大学大学院医学系研究科 准教授.
片桐 秀樹(Hideki Katagiri)
東北大学大学院医学系研究科 教授.
研究室URL:http://www.atmd.med.tohoku.ac.jp/
© 2013 突田壮平・山田哲也・片桐秀樹 Licensed under CC 表示 2.1 日本
(東北大学大学院医学系研究科 附属創生応用医学研究センター代謝疾患学分野)
email:突田壮平,山田哲也
DOI: 10.7875/first.author.2013.003
Hepatic glucokinase modulates obesity predisposition by regulating BAT thermogenesis via neural signals.
Sohei Tsukita, Tetsuya Yamada, Kenji Uno, Kei Takahashi, Keizo Kaneko, Yasushi Ishigaki, Junta Imai, Yutaka Hasegawa, Shojiro Sawada, Hisamitsu Ishihara, Yoshitomo Oka, Hideki Katagiri
Cell Metabolism, 16, 825-832 (2012)
要 約
体重は本質的には摂取エネルギーと消費エネルギーとの収支により決定されるが,日々の食事量や活動量の変動がそのまま直接的に体重の変化に反映されるわけではない.近年,脳をオーガナイザーとする多くの臓器間相互作用がフィーバック機構として機能し,過剰なエネルギーの摂取に対する体重の恒常性の維持に寄与していることが明らかになった.一方,飽食の現代において肥満人口が増加の一途をたどっていることから,エネルギーの摂取に対しフィードフォワード機構としてはたらき体重の増加に寄与する臓器間相互作用の存在が想定されるものの,その存在は明らかでなかった.この研究では,この一端を説明しうる,肝臓からの新たな臓器間神経ネットワークを見い出した.エネルギーの摂取過剰の条件において,グルコキナーゼの発現上昇にともない肝臓における糖代謝に変化が生じると,その変化により惹起されたシグナルが迷走神経の求心路を介し伝達される.これをうけ,延髄から褐色脂肪組織へといたる交感神経の活性が低下し,褐色脂肪組織における熱産生が抑制され脂肪の蓄積が促進される.この機構はエネルギーの摂取増加に呼応するエネルギー消費の低下をつかさどっており,その観点から個体レベルにおける倹約機構と考えられた.食事を満足に得ることのできなかった時代において生存に有利にはたらいたと考えられるこの機構は,皮肉にも,飽食の現代において肥満に舵を切る分子機構としてはたらいているものと想定された.さらに,この機構のはたらきの違いが,肥満のなりやすさに関するマウスの系統の違いを規定する一因となっていることが示された.
はじめに
肥満は糖尿病,高脂血症,高血圧症など生活習慣病に共通する要因であり,その点からも肥満の研究,すなわち,体重の制御機構の解明は重要である.体重の恒常性を維持することの本質的な意義は,食物をつねに得ることができるかどうか不確かな環境において体にすぐに利用できるエネルギー源を蓄えておくことにあったと考えられる.一方で,脂肪の過剰な蓄積は個体の活動性を損ない捕食や外敵からの回避において不利益をもたらすことになる.したがって,エネルギー源の貯蓄量と補給量とにもとづき摂食行動およびエネルギー消費量を適正に制御する必要があり,視床下部はこの分子機構において中心的な役割を担っている.たとえば,蓄積する中性脂肪の量に応じ白色脂肪細胞から分泌されるレプチンは,視床下部に作用し摂食の抑制およびエネルギー消費の亢進をもたらすことによりエネルギーの摂取過剰に対するフィードバック機構を構成することが知られている1).また,レプチンのような液性因子にくわえ,近年,神経ネットワークもエネルギー代謝の恒常性の維持に重要な役割をはたしていることが明らかになってきた2).いずれにしても,エネルギー摂取の増加に対し体重の恒常性を維持するよう機能するフィードバック機構が厳格に機能するならば体重の増加は起こりえず,したがって,肥満は生じないはずである.しかしながら,肥満人口が増加の一途をたどっている状況を鑑みると,体重の恒常性の維持の機構は容易に破綻する,もしくは,体重の増加にむけ機能するフィードフォワード機構の存在が想起される.
筆者らは,後者の観点から,肝臓における糖代謝の変化が全身のエネルギー代謝に及ぼす影響を検討することにした.なぜなら,肝臓において特異的にグルコキナーゼを過剰発現するトランスジェニックマウスは肥満の表現型を示すことが報告されており3),肝臓における糖代謝が全身のエネルギー代謝に影響を及ぼすことが示唆されていたからである.グルコキナーゼはインスリンにより強力に発現誘導され,肝細胞に取り込まれたグルコースをグルコース6-リン酸に変換する反応を触媒する酵素である.この研究では,アデノウイルス遺伝子発現系を用いてマウスの肝臓において選択的にグルコキナーゼを過剰発現させることにより肝臓における解糖系を亢進し,肝臓における糖代謝の変化が全身のエネルギー収支,とくに,褐色脂肪組織におけるエネルギー消費に及ぼす影響を検討した.褐色脂肪組織は熱産生タンパク質であるUCP1を特異的に発現し,酸化的リン酸化を脱共役させることによりエネルギーを熱として消費するという機能をもつ.これまで,褐色脂肪組織はげっ歯類やヒトの新生児に特異的に存在するものとされていたが,最近,ヒトの成人においても機能的に活発な褐色脂肪組織の存在することや,褐色脂肪組織の量や活性が低いと肥満につながる可能性のあることが報告され,注目をあつめている4).
1.肝臓におけるグルコキナーゼの発現上昇はグリコーゲンの蓄積を増加させる
C57BL/6マウスに高脂肪かつ高スクロース食を負荷したところ,肝臓における内因性のグルコキナーゼの発現は負荷1週目より著明に上昇した.そこで,この負荷の早期より生じる肝臓におけるグルコキナーゼの発現上昇に着目し,個体レベルでのエネルギー代謝に及ぼす影響を調べることにした.通常食により飼育しているマウスの肝臓にアデノウイルスベクターを用いてグルコキナーゼを過剰発現させたところ,その発現量に依存して肝臓におけるグリコーゲンおよび中性脂肪の量が増加した.マイクロアレイを用いた遺伝子発現解析ではグリコーゲン合成および脂肪合成に関与する酵素の遺伝子の発現上昇が確認された.このマウスでは,対照となる野生型マウスと比較して,摂餌量および活動量に差は認められなかった.また,プローブを用いて直接的に測定した肝臓の深部の温度にも差は認められず,赤外線サーモグラフィー画像においても肝臓の表面の温度に差は認められなかった.これらのことより,グルコキナーゼの発現上昇は肝臓における熱産生には影響を及ぼさないことが判明した.
2.肝臓におけるグルコキナーゼの発現は交感神経の活性を下方制御することにより褐色脂肪組織における熱産生を抑制する
肝臓においてグルコキナーゼを過剰発現させたマウスにおいて,肩甲骨のあいだに存在する褐色脂肪組織の組織像を検討したところ,興味深いことに,脂肪滴の直径が増加しており熱産生の低下が示唆された.また,肝臓におけるグルコキナーゼの発現量に依存して,褐色脂肪組織における熱産生関連遺伝子UCP1遺伝子,PGC-1α遺伝子,D2遺伝子の発現が低下しており,これは脂肪滴の直径の増大と相応した.これら熱産生遺伝子は交感神経系の活性によりその発現が制御されていることが知られており,交感神経系の活性の低下が強く示唆された.実際に,褐色脂肪組織におけるノルアドレナリンの代謝回転を測定したところ低下しており,褐色脂肪組織にいたる交感神経の活性が低下していることが確認された.さらに,褐色脂肪組織において熱産生を制御する交感神経のプレモーターニューロンが存在する延髄の吻側の縫線核において5),神経活性化のマーカーであるc-fosはmRNAレベルおよびタンパク質レベルにおいて低下していた.これらのことより,肝臓におけるグルコキナーゼの発現上昇は,少なくとも,延髄のプレモーターニューロンのレベルにおいて交感神経の活性を下方制御し,褐色脂肪組織における熱産生を抑制しているものと考えられた.
褐色脂肪組織における熱産生の低下が個体レベルでのエネルギー消費に及ぼす影響を,ノルアドレナリンの投与に対する酸素消費量の増加量(適応性非ふるえ熱産生能6))により評価した.肝臓においてグルコキナーゼを過剰発現させたマウスではノルアドレナリンの投与に対する酸素消費量の増加反応が低下しており,個体レベルでのエネルギー消費の低下していることが確認された.さらに,このマウスを温度的中性条件である28℃において飼育すると,肝臓におけるグルコキナーゼの発現量に依存して体重および白色脂肪の重量は増加した.以上より,肝臓におけるグルコキナーゼの発現上昇により褐色脂肪組織における熱産生が低下し,個体レベルでのエネルギー消費が減少し体重および脂肪量の増加することが示された.
3.肝臓の発する迷走神経のシグナルが褐色脂肪組織における熱産生を抑制する
肝臓におけるグルコキナーゼの発現上昇は交感神経の活性を下方制御し褐色脂肪組織における熱産生を抑制するわけだが,シグナルはどのような経路を介し肝臓から脳へと伝達されるのだろうか? 肝臓における代謝のシグナルが迷走神経の求心路を介し中枢へと伝達されることが報告されていたことから7,8),肝臓と中枢とをつなぐ経路として迷走神経の求心路の関与を考えた.迷走神経の肝臓枝を選択的に切断したのち肝臓においてグルコキナーゼを過剰発現させたところ,迷走神経を切断しないマウスで観察された褐色脂肪組織における脂肪滴の直径の増大,熱産生関連遺伝子の発現低下,縫線核におけるc-fosの発現低下などの変化は認められなくなった.このことから,褐色脂肪組織における熱産生の抑制は肝臓の発する迷走神経のシグナルを介すものと考えられた(図1).
4.肝臓と褐色脂肪組織との組織間の機構はレプチンによるエネルギー消費の亢進作用を抑制する
レプチンは視床下部に作用し摂食の抑制およびエネルギー消費の亢進をもたらすことにより,エネルギーの摂取過剰に対しフィードバック機構としてはたらき,体重の恒常性の維持に重要な役割を担うことが知られている.そこで,レプチンによるフィードバック機構と肝臓と褐色脂肪組織とのあいだのフィードフォワード機構との相互作用について検討した.肝臓においてグルコキナーゼを過剰発現させたマウスの腹腔にレプチンを投与したところ,対照となる野生型マウスと同等の摂食の抑制効果が認められた.さらに,この両方のマウスにおいて,レプチンの投与により視床下部の弓状核におけるc-fosの発現上昇,摂食関連ペプチドのうち摂食抑制作用をもつPOMCの発現上昇および摂食亢進作用をもつNPYの発現抑制が認められた.以上より,肝臓のグルコキナーゼにより誘導される迷走神経のシグナルは,レプチンによる摂食の抑制効果に影響をあたえないことが判明した.一方,レプチンの投与により野生型マウスにおいて認められた縫線核におけるc-fos発現上昇や褐色脂肪組織における熱産生関連遺伝子の発現上昇は,肝臓においてグルコキナーゼを過剰発現させたマウスにおいて認められなかった.したがって,この迷走神経のシグナルは,縫線核-交感神経-褐色脂肪組織とつながる熱産生経路を抑制するものと考えられた.さらに,縫線核の上流において褐色脂肪組織へといたる交感神経の活性の制御に関与する視床下部の室傍核9) に対する,レプチンの投与による活性化は,肝臓においてグルコキナーゼを過剰発現させたマウスにおいても影響をうけなったことから,このシグナルはレプチンによるエネルギー消費の亢進作用を視床下部より下流において抑制するものと考えられた(図2).
5.肝臓におけるグルコキナーゼの発現は肥満になりやすさを規定する
肝臓におけるグルコキナーゼの発現上昇に端を発する肝臓と褐色脂肪組織との組織間の機構は,病態生理的にはどのような役割をはたしているのだろうか? 過剰なエネルギーの摂取により肥満をきたしやすいC57BL/6マウスおよびAKRマウスと,肥満をきたしにくいSWR/JマウスおよびA/Jマウスに対し10),高脂肪かつ高スクロース食を1週間にわたり負荷し,肝臓における内因性のグルコキナーゼの発現および褐色脂肪組織におけるUCP1遺伝子の発現を比較した.高脂肪かつ高スクロース食の負荷により,肝臓におけるグルコキナーゼの発現は,肥満をきたしやすい系統のマウスにおいて顕著に上昇した.一方,褐色脂肪組織におけるUCP1遺伝子の発現は,肥満をきたしにくい系統のマウスにおいて上昇した.すなわち,肝臓におけるグルコキナーゼの発現と褐色脂肪組織におけるUCP1遺伝子の発現のあいだには逆相関が認められた.
肝臓と褐色脂肪組織との組織間の機構が肥満の指向性に関与している可能性を考え,以下の実験を行った.まず,肥満をきたしにくい系統であるSWR/Jマウスの肝臓においてグルコキナーゼを過剰発現させたところ,褐色脂肪組織における熱産生は低下し体重の増加が促進された.つぎに,肥満をきたしやすい系統であるC57BL/6マウスにおいて脂肪かつ高スクロース食の負荷により生じる肝臓における内因性のグルコキナーゼの発現上昇に対しこれをshRNAによりノックダウンしたところ,褐色脂肪組織における熱産生は増加し体重の増加は抑制された.以上より,高脂肪かつ高スクロース食の負荷にともなう肝臓における内因性のグルコキナーゼの発現上昇は,熱産生能を制御することにより肥満の指向性を決める一因となると考えられた.
おわりに
この研究により,これまで見い出されていなかった,エネルギーの過剰な摂取に対する体重制御におけるフィードフォワード機構の存在が明らかになった.食物の供給が不安定な自然環境では,余分に摂取したエネルギーを脂肪として効率よく蓄積することは飢餓に直面した際に個体の生存に有利にはたらくと思われ,その観点から,この機構は個体レベルでの“倹約機構”と考えられる.一方,現代社会のような慢性的に過栄養な環境においてこの機構は体重増加のきっかけとなり,ひいては,肥満の発症に関与するものと考えられる.
今後の課題として,肝臓における糖代謝の変化がどのような分子機構を介し迷走神経の活動を変化させるのか,ヒトの成人においてもこの倹約機構が機能しているか,など明らかにすべき点は多い.このフィードフォワード機構に介入し,肥満者において低下している褐色脂肪組織における熱産生を活性化することができれば,過栄養時代における新たな肥満症の治療法や予防法の確立につながるものと考えられる.
文 献
- Rosen, E. D. & Spiegelman, B. M.: Adipocytes as regulators of energy balance and glucose homeostasis. Nature, 444, 847-53 (2006)[PubMed]
- Katagiri, H., Yamada, T. & Oka, Y.: Adiposity and cardiovascular disorders: disturbance of the regulatory system consisting of humoral and neuronal signals. Circ. Res., 101, 27-39 (2007)[PubMed]
- Ferre, T., Riu, E., Franckhauser, S. et al.: Long-term overexpression of glucokinase in the liver of transgenic mice leads to insulin resistance. Diabetologia, 46, 1662-1668 (2003)[PubMed]
- Cypess, A. M., Lehman, S., Williams, G. et al.: Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med., 360, 1509-1517 (2009)[PubMed]
- Morrison, S. F., Nakamura, K. & Madden, C. J.: Central control of thermogenesis in mammals. Exp. Physiol., 93, 773-797 (2008)[PubMed]
- Golozoubova, V., Cannon, B. & Nedergaard, J.: UCP1 is essential for adaptive adrenergic nonshivering thermogenesis. Am. J. Physiol. Endocrinol. Metab., 291, E350-E357 (2006)[PubMed]
- Bernal-Mizrachi, C., Xiaozhong, L., Yin, L. et al.: An afferent vagal nerve pathway links hepatic PPARα activation to glucocorticoid-induced insulin resistance and hypertension. Cell Metab., 5, 91-102 (2007)[PubMed]
- Uno, K., Katagiri, H., Yamada, T. et al.: Neuronal pathway from the liver modulates energy expenditure and systemic insulin sensitivity. Science, 312, 1656-1659 (2006)[PubMed]
- Morris, D. L. & Rui, L.: Recent advances in understanding leptin signaling and leptin resistance. Am. J. Physiol. Endocrinol. Metab., 297, E1247-E1259 (2009)[PubMed]
- West, D. B., Boozer, C. N., Moody, D. L. et al.: Dietary obesity in nine inbred mouse strains. Am. J. Physiol., 262, R1025-R1032 (1992)[PubMed]
著者プロフィール
略歴:2011年 東北大学大学院医学系研究科 修了,同年より同 助手.
研究テーマ:糖代謝およびエネルギー代謝の制御機構.
抱負:糖尿病や肥満症の病態の解明また治療法の開発に貢献したい.日常の診療では体重増加を助長しない血糖降下の手助けをしたい.
山田 哲也(Tetsuya Yamada)
東北大学大学院医学系研究科 准教授.
片桐 秀樹(Hideki Katagiri)
東北大学大学院医学系研究科 教授.
研究室URL:http://www.atmd.med.tohoku.ac.jp/
© 2013 突田壮平・山田哲也・片桐秀樹 Licensed under CC 表示 2.1 日本
