ピロリ菌のもつがんタンパク質CagAは酸化ストレスにより誘導されるオートファジーにより分解される
津川 仁・鈴木秀和
(慶應義塾大学医学部 内科学教室消化器内科)
email:津川 仁,鈴木秀和
DOI: 10.7875/first.author.2012.162
Reactive oxygen species-induced autophagic degradation of Helicobacter pylori CagA is specifically suppressed in cancer stem-like cells.
Hitoshi Tsugawa, Hidekazu Suzuki, Hideyuki Saya, Masanori Hatakeyama, Toshiya Hirayama, Kenro Hirata, Osamu Nagano, Juntaro Matsuzaki, Toshifumi Hibi
Cell Host & Microbe, 12, 764-777 (2012)
ピロリ菌のもつエフェクタータンパク質CagAはIV型分泌装置により宿主細胞へ移行しがんタンパク質として機能する.したがって,宿主細胞におけるCagAの安定性は,ピロリ菌に感染した際の発がんリスクを規定する重要な要因であると考えられる.この研究において,筆者らは,宿主細胞に移行したCagAは,やはりピロリ菌により産生される分泌毒素VacAを介した活性酸素種の増加により誘導されるオートファジーにより分解されることを示した.通常,宿主細胞に移行したCagAはオートファジーにより排除されるため長期的に安定して存在することはない.しかし,CD44v9を発現するがん幹細胞では,VacAにより誘導される活性酸素種の蓄積が生じないためオートファジーが起こらず,CagAが特異的に蓄積することがわかった.つまり,ピロリ菌に感染した際のCD44v9陽性がん幹細胞の存在は,胃がんの発がんリスクを亢進させるものと考えられた.この研究により,ピロリ菌と胃がんとの関係が,ピロリ菌とCD44v9陽性がん幹細胞との相互作用から直接的に証明され,胃がんの予防および治療の標的としてのCD44v9陽性がん幹細胞の重要性が示された.
ピロリ菌(Helicobacter pylori)のゲノムにはcagPAI(cag pathogenicity island)とよばれる約40 kbの起源の不明な領域が存在し,cagPAIによりコードされるCagAは,同じくcagPAIにコードされ外膜に発現する注射針様の構造体であるIV型分泌装置により宿主細胞へと導入される.宿主細胞に侵入したCagAは,がんタンパク質SHP2や極性制御タンパク質PAR1との相互作用により,異常な細胞増殖シグナルの惹起および上皮細胞の極性破壊を誘導する1,2).さらに,トランスジェニックマウスを用いた解析によりCagAはマウスに対し単独で発がん活性を示すことが明らかになり,CagAは細菌に由来するがんタンパク質であることが証明されている3).
CagAがSHP2やPAR1との相互作用を介し発がん活性を持続するためには移行した宿主細胞において安定して存在しなければならないが,はたして細菌に由来する異物タンパク質が哺乳動物の細胞において安定して存在できるのかというのが,この研究における最初の疑問であった.当時,宿主細胞におけるCagAの半減期は約200分であることが報告されており4),筆者らによる検討でも,宿主細胞に移行したCagAは時間に依存して分解されることが確認された.つまり,宿主細胞は異物タンパク質であるCagAに対する排除応答を惹起しており,CagAの安定な存在をゆるしていないと考えられた.一方で,宿主細胞の惹起するCagA排除機構が遅延もしくは破綻した細胞ではCagAが特異的に蓄積し,このような細胞の存在はピロリ菌に感染した際の発がんリスクを亢進させるとともに,発がんリスクを規定する要因ともなると考えられた.この研究では,ピロリ菌に感染した細胞の示すCagA排除応答の詳細を検討し,その結果にもとづき,CagA排除応答の破綻によりCagAの蓄積を示した細胞の性質を調べた.
ピロリ菌から宿主細胞に移行したCagA,および,宿主細胞においてSrcファミリーキナーゼによりチロシン残基がリン酸化されたリン酸化CagAは,ともに時間に依存して分解されることが示された.このことから,宿主細胞に移行したCagAは安定して存在することはできず,宿主細胞においてCagAに対する排除応答が発動されているものと考えられた.そこで,このCagAの排除機構について調べた.オートファジー阻害剤である3-メチルアデニンおよびウォルトマンニンはCagAおよびリン酸化CagAの分解を有意に抑制し,また,mTORの阻害によるオートファジー誘導剤であるラパマイシンをCagAを強制発現させた細胞に処理したところCagAの分解は促進された.さらに,免疫電子顕微鏡法による解析によりオートファゴソームにおいてCagAのシグナルが認められた.これらの結果から,宿主細胞に移行したCagAはオートファジーによる分解をうけるものと結論づけられた.
ピロリ菌に感染した細胞ではオートファジーが誘導されることが報告されている5).筆者らも,これを確認した.興味深いことに,CagAを強制発現させた細胞ではオートファジーは誘導されなかったことから,CagAの分解にはたらくオートファジーはCagA以外のピロリ菌に由来する因子により誘導されると考えられた.近年,ピロリ菌のもつ分泌型タンパク質毒素であるVacAが宿主細胞に対しオートファジーを誘導することが報告され5,6),くわえて最近,筆者らは,このオートファジーの誘導の際のVacAの受容体はLRP1であることを報告した7).VacAの構造は,宿主細胞における空胞の形成に重要なN末端側の断片(p33断片)と,受容体の認識に機能するp55断片からなる.p55断片にはmid-regionとよばれる標的となる細胞への結合に重要な領域が含まれ,VacAはこの領域における遺伝子配列の違いからm1型VacAとm2型VacAとに大別される.m1型VacAとm2型VacAとで標的となる細胞に対する特異性に差異が認められることから,この領域が受容体の認識にとり重要であることが強く示唆されている8).
m1型VacAはCagA分解性オートファジーに対する誘導活性を示したのに対し,m2型VacAは誘導活性を示さなかった.さらに,LRP1をノックダウンした細胞ではCagA分解性オートファジーは誘導されずCagAは蓄積した.また,オートファジーの誘導活性を示さないm2型VacAはLRP1との結合能をもたないことも明らかになった.つまり,m1型VacAのLRP1への結合は,CagA分解性オートファジーの誘導における重要な初期反応であると考えられた.
それでは,VacAはLRP1への結合を介してどのようなシグナルを惹起しCagA分解性オートファジーを誘導するのだろうか? m1型VacAを産生するピロリ菌に感染した細胞には活性酸素種の蓄積が認められるのに対し,m2型VacAを産生するピロリ菌に感染した細胞には蓄積は認められなかった.活性酸素種はオートファジーを惹起する因子のひとつであることが知られていることから,m1型VacAに依存した活性酸素種の蓄積はCagA分解性オートファジーの誘導に寄与しているかどうか検討した.その結果,ピロリ菌に感染した細胞に抗酸化剤N-アセチルシステインを処理することによりオートファジーの誘導は阻害されCagAの分解も抑制された.一方で,NADPHオキシダーゼ阻害剤やMnスーパーオキシドジスムターゼによる処理ではオートファジーの誘導阻害およびCagAの蓄積は認められなかった.これらの結果から,CagA分解性オートファジーの誘導に活性酸素種の蓄積は寄与するが,その起源はNADPHオキシダーゼあるいはミトコンドリアに由来するものではないと考えられた.
N-アセチルシステインはシステインのプロドラッグであることから,ピロリ菌に感染した際のグルタチオンの減少が活性酸素種の蓄積を介しオートファジーの誘導につながるとの仮説をたて検討した.その結果,ピロリ菌に感染した細胞においてグルタチオンは有意に減少し,また,CagAを強制発現させた細胞および胃がん上皮細胞であるAGS細胞においてグルタチオンはm1型VacAの用量に依存して減少した.また,オートファジー誘導活性のないm2型VacAはグルタチオンを減少させなかった.さらに,ピロリ菌に感染した細胞では生存シグナルのひとつであるAktのThr308およびSer473のリン酸化が亢進しており,これらはCagAに依存しないこと,また,Ser473のリン酸化は活性酸素種の蓄積に依存していることが明らかになった.さらに,Aktのリン酸化はp53に特異的なユビキチンリガーゼであるMdm2のリン酸化を誘導し,p53のタンパク質分解は亢進した.p53の減少はオートファジーを誘導することが報告されており9),これらの結果から,m1型VacAはLRP1への結合ののち,グルタチオンを減少させることにより活性酸素種が蓄積し,それによりAktのリン酸化が亢進してMdm2が活性化され,p53の分解および減少によりCagA分解性オートファジーが起こると結論づけられた(図1).
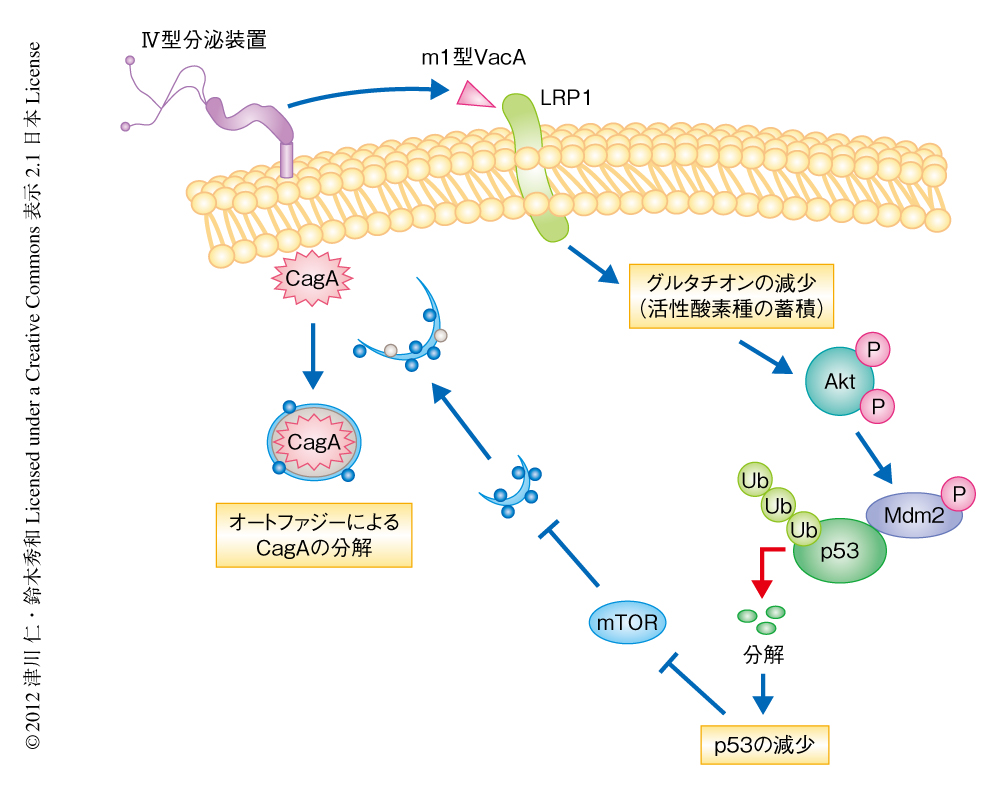
VacAによるオートファジーの誘導を介した宿主細胞におけるCagAの分解は“ピロリ菌に感染した際の胃がんの発症へのCagAの寄与”に異議をとなえるものだと考えられるが,オートファジーの誘導に阻害を示す細胞ではCagAが特異的に蓄積し発がんリスクが高まるのではないかとも推測できる.がん幹細胞は,がん組織に存在してがん細胞を生み出し,また,抗がん剤や放射線治療に対する抵抗性およびがんの再発や転移に寄与する.近年,がん幹細胞の主要な表面マーカータンパク質のひとつであるCD44バリアント型CD44v9は,活性酸素種の蓄積を防ぎ腫瘍の増大や抗がん剤への抵抗性に寄与することが明らかにされている10).CD44v9陽性がん幹細胞は,細胞膜においてシスチントランスポーターであるxCTを安定化させ,グルタチオンを増加させることにより活性酸素種の蓄積を防いでいることが明らかにされている10).
CD44v9陽性がん幹細胞ではm1型VacAによるグルタチオンの減少に阻害を示し,CagA分解性オートファジーが起こらずCagAが蓄積するのではないかと仮説をたてた.そこで,CD44陰性のMKN28細胞に通常型のCD44sおよびCD44v9をそれぞれ強制発現させたのち,ピロリ菌に感染した細胞におけるグルタチオンのレベルを検討した.CD44sを強制発現させた細胞とは対照的に,CD44v9を強制発現させた細胞ではピロリ菌に感染してもグルタチオンは減少しなかった.その結果,AktのSer473のリン酸化,Mdm2のリン酸化,p53の減少は誘導されず,オートファジーの誘導は抑制されCagAの蓄積が認められた.また,CD44v9を強制発現させたMKN28細胞にピロリ菌を感染させxCTの阻害剤であるスルファサラジンを処理すると,Aktのリン酸化およびMdm2のリン酸化が誘導されp53は減少し,CagA分解性オートファジーの誘導とともにCagAの分解が生じた.これらの結果から,CD44v9を発現する細胞におけるCagAの蓄積は,CD44v9によるxCTの安定化を介したグルタチオンの増加に依存していることが示された(図2).
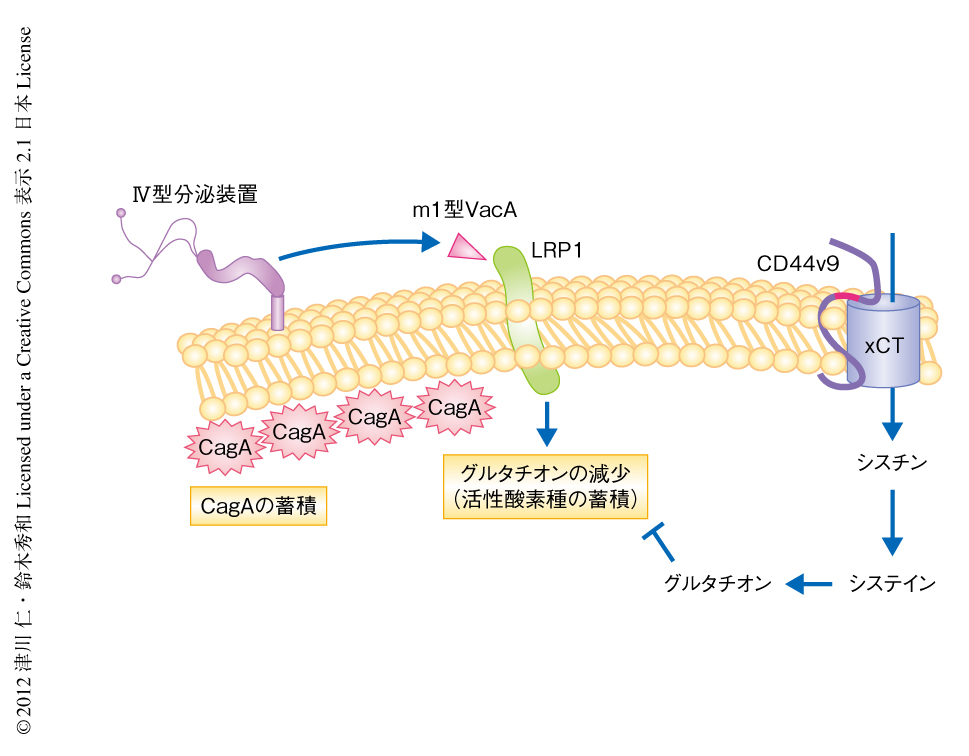
宿主細胞に移行したCagAはSHP2およびPAR1との相互作用を介し異常な細胞増殖を惹起し上皮細胞の極性を破壊する.そのため,宿主細胞におけるCagAの過剰な安定は上皮細胞の脱落を容易に誘発し,ピロリ菌にとり,定着した足場の喪失につながる.そこで,ピロリ菌は胃における長期的な生存戦略のひとつとして,VacAによるオートファジーの誘導を介しCagAを負に制御する能力を獲得したとも考えられる.一方で,CD44v9陽性がん幹細胞はVacAによるCagA分解性オートファジーの誘導に阻害を示すことによりCagAを蓄積する.したがって,CD44v9陽性がん幹細胞の存在はピロリ菌に感染した際の胃がんの発がんに強く関与し,また,胃がんの発がんリスクを亢進させる要因になると考えられる.この研究により,ピロリ菌とCD44v9陽性がん幹細胞との遭遇が,ヒトにとりピロリ菌に感染した際の胃がんの発がんリスクを規定する要因となることが示され,これまでの機序では説明できなかった,ピロリ菌の感染者すべてが胃がんを発症するのではないという事実の一部を説明することになった.今後の胃がんの予防および治療の標的として,ピロリ菌の感染という根本的な標的はいうまでもなく,長期の炎症を背景に出現すると考えられるCD44v9陽性がん幹細胞についてもその重要性を提示することができたわけである.
略歴:2007年 東北薬科大学大学院薬学研究科博士課程 修了,同年 慶應義塾大学医学部 共同研究員を経て,同年より同 特任助教.
研究テーマ:ピロリ菌の感染の分子機構と発がん.
関心事:細菌と宿主との攻防戦と,その結果,生じる病態の解明.
鈴木 秀和(Hidekazu Suzuki)
慶應義塾大学医学部 准教授.
© 2012 津川 仁・鈴木秀和 Licensed under CC 表示 2.1 日本
(慶應義塾大学医学部 内科学教室消化器内科)
email:津川 仁,鈴木秀和
DOI: 10.7875/first.author.2012.162
Reactive oxygen species-induced autophagic degradation of Helicobacter pylori CagA is specifically suppressed in cancer stem-like cells.
Hitoshi Tsugawa, Hidekazu Suzuki, Hideyuki Saya, Masanori Hatakeyama, Toshiya Hirayama, Kenro Hirata, Osamu Nagano, Juntaro Matsuzaki, Toshifumi Hibi
Cell Host & Microbe, 12, 764-777 (2012)
要 約
ピロリ菌のもつエフェクタータンパク質CagAはIV型分泌装置により宿主細胞へ移行しがんタンパク質として機能する.したがって,宿主細胞におけるCagAの安定性は,ピロリ菌に感染した際の発がんリスクを規定する重要な要因であると考えられる.この研究において,筆者らは,宿主細胞に移行したCagAは,やはりピロリ菌により産生される分泌毒素VacAを介した活性酸素種の増加により誘導されるオートファジーにより分解されることを示した.通常,宿主細胞に移行したCagAはオートファジーにより排除されるため長期的に安定して存在することはない.しかし,CD44v9を発現するがん幹細胞では,VacAにより誘導される活性酸素種の蓄積が生じないためオートファジーが起こらず,CagAが特異的に蓄積することがわかった.つまり,ピロリ菌に感染した際のCD44v9陽性がん幹細胞の存在は,胃がんの発がんリスクを亢進させるものと考えられた.この研究により,ピロリ菌と胃がんとの関係が,ピロリ菌とCD44v9陽性がん幹細胞との相互作用から直接的に証明され,胃がんの予防および治療の標的としてのCD44v9陽性がん幹細胞の重要性が示された.
はじめに
ピロリ菌(Helicobacter pylori)のゲノムにはcagPAI(cag pathogenicity island)とよばれる約40 kbの起源の不明な領域が存在し,cagPAIによりコードされるCagAは,同じくcagPAIにコードされ外膜に発現する注射針様の構造体であるIV型分泌装置により宿主細胞へと導入される.宿主細胞に侵入したCagAは,がんタンパク質SHP2や極性制御タンパク質PAR1との相互作用により,異常な細胞増殖シグナルの惹起および上皮細胞の極性破壊を誘導する1,2).さらに,トランスジェニックマウスを用いた解析によりCagAはマウスに対し単独で発がん活性を示すことが明らかになり,CagAは細菌に由来するがんタンパク質であることが証明されている3).
CagAがSHP2やPAR1との相互作用を介し発がん活性を持続するためには移行した宿主細胞において安定して存在しなければならないが,はたして細菌に由来する異物タンパク質が哺乳動物の細胞において安定して存在できるのかというのが,この研究における最初の疑問であった.当時,宿主細胞におけるCagAの半減期は約200分であることが報告されており4),筆者らによる検討でも,宿主細胞に移行したCagAは時間に依存して分解されることが確認された.つまり,宿主細胞は異物タンパク質であるCagAに対する排除応答を惹起しており,CagAの安定な存在をゆるしていないと考えられた.一方で,宿主細胞の惹起するCagA排除機構が遅延もしくは破綻した細胞ではCagAが特異的に蓄積し,このような細胞の存在はピロリ菌に感染した際の発がんリスクを亢進させるとともに,発がんリスクを規定する要因ともなると考えられた.この研究では,ピロリ菌に感染した細胞の示すCagA排除応答の詳細を検討し,その結果にもとづき,CagA排除応答の破綻によりCagAの蓄積を示した細胞の性質を調べた.
1.宿主細胞に移行したCagAはオートファジーにより分解される
ピロリ菌から宿主細胞に移行したCagA,および,宿主細胞においてSrcファミリーキナーゼによりチロシン残基がリン酸化されたリン酸化CagAは,ともに時間に依存して分解されることが示された.このことから,宿主細胞に移行したCagAは安定して存在することはできず,宿主細胞においてCagAに対する排除応答が発動されているものと考えられた.そこで,このCagAの排除機構について調べた.オートファジー阻害剤である3-メチルアデニンおよびウォルトマンニンはCagAおよびリン酸化CagAの分解を有意に抑制し,また,mTORの阻害によるオートファジー誘導剤であるラパマイシンをCagAを強制発現させた細胞に処理したところCagAの分解は促進された.さらに,免疫電子顕微鏡法による解析によりオートファゴソームにおいてCagAのシグナルが認められた.これらの結果から,宿主細胞に移行したCagAはオートファジーによる分解をうけるものと結論づけられた.
2.CagAの分解にはたらくオートファジーはピロリ菌により産生される分泌毒素VacAにより誘導される
ピロリ菌に感染した細胞ではオートファジーが誘導されることが報告されている5).筆者らも,これを確認した.興味深いことに,CagAを強制発現させた細胞ではオートファジーは誘導されなかったことから,CagAの分解にはたらくオートファジーはCagA以外のピロリ菌に由来する因子により誘導されると考えられた.近年,ピロリ菌のもつ分泌型タンパク質毒素であるVacAが宿主細胞に対しオートファジーを誘導することが報告され5,6),くわえて最近,筆者らは,このオートファジーの誘導の際のVacAの受容体はLRP1であることを報告した7).VacAの構造は,宿主細胞における空胞の形成に重要なN末端側の断片(p33断片)と,受容体の認識に機能するp55断片からなる.p55断片にはmid-regionとよばれる標的となる細胞への結合に重要な領域が含まれ,VacAはこの領域における遺伝子配列の違いからm1型VacAとm2型VacAとに大別される.m1型VacAとm2型VacAとで標的となる細胞に対する特異性に差異が認められることから,この領域が受容体の認識にとり重要であることが強く示唆されている8).
m1型VacAはCagA分解性オートファジーに対する誘導活性を示したのに対し,m2型VacAは誘導活性を示さなかった.さらに,LRP1をノックダウンした細胞ではCagA分解性オートファジーは誘導されずCagAは蓄積した.また,オートファジーの誘導活性を示さないm2型VacAはLRP1との結合能をもたないことも明らかになった.つまり,m1型VacAのLRP1への結合は,CagA分解性オートファジーの誘導における重要な初期反応であると考えられた.
3.CagAの分解にはたらくオートファジーは宿主細胞における活性酸素種の蓄積を介し誘導される
それでは,VacAはLRP1への結合を介してどのようなシグナルを惹起しCagA分解性オートファジーを誘導するのだろうか? m1型VacAを産生するピロリ菌に感染した細胞には活性酸素種の蓄積が認められるのに対し,m2型VacAを産生するピロリ菌に感染した細胞には蓄積は認められなかった.活性酸素種はオートファジーを惹起する因子のひとつであることが知られていることから,m1型VacAに依存した活性酸素種の蓄積はCagA分解性オートファジーの誘導に寄与しているかどうか検討した.その結果,ピロリ菌に感染した細胞に抗酸化剤N-アセチルシステインを処理することによりオートファジーの誘導は阻害されCagAの分解も抑制された.一方で,NADPHオキシダーゼ阻害剤やMnスーパーオキシドジスムターゼによる処理ではオートファジーの誘導阻害およびCagAの蓄積は認められなかった.これらの結果から,CagA分解性オートファジーの誘導に活性酸素種の蓄積は寄与するが,その起源はNADPHオキシダーゼあるいはミトコンドリアに由来するものではないと考えられた.
N-アセチルシステインはシステインのプロドラッグであることから,ピロリ菌に感染した際のグルタチオンの減少が活性酸素種の蓄積を介しオートファジーの誘導につながるとの仮説をたて検討した.その結果,ピロリ菌に感染した細胞においてグルタチオンは有意に減少し,また,CagAを強制発現させた細胞および胃がん上皮細胞であるAGS細胞においてグルタチオンはm1型VacAの用量に依存して減少した.また,オートファジー誘導活性のないm2型VacAはグルタチオンを減少させなかった.さらに,ピロリ菌に感染した細胞では生存シグナルのひとつであるAktのThr308およびSer473のリン酸化が亢進しており,これらはCagAに依存しないこと,また,Ser473のリン酸化は活性酸素種の蓄積に依存していることが明らかになった.さらに,Aktのリン酸化はp53に特異的なユビキチンリガーゼであるMdm2のリン酸化を誘導し,p53のタンパク質分解は亢進した.p53の減少はオートファジーを誘導することが報告されており9),これらの結果から,m1型VacAはLRP1への結合ののち,グルタチオンを減少させることにより活性酸素種が蓄積し,それによりAktのリン酸化が亢進してMdm2が活性化され,p53の分解および減少によりCagA分解性オートファジーが起こると結論づけられた(図1).
4.CagAはCD44v9陽性がん幹細胞において特異的に蓄積する
VacAによるオートファジーの誘導を介した宿主細胞におけるCagAの分解は“ピロリ菌に感染した際の胃がんの発症へのCagAの寄与”に異議をとなえるものだと考えられるが,オートファジーの誘導に阻害を示す細胞ではCagAが特異的に蓄積し発がんリスクが高まるのではないかとも推測できる.がん幹細胞は,がん組織に存在してがん細胞を生み出し,また,抗がん剤や放射線治療に対する抵抗性およびがんの再発や転移に寄与する.近年,がん幹細胞の主要な表面マーカータンパク質のひとつであるCD44バリアント型CD44v9は,活性酸素種の蓄積を防ぎ腫瘍の増大や抗がん剤への抵抗性に寄与することが明らかにされている10).CD44v9陽性がん幹細胞は,細胞膜においてシスチントランスポーターであるxCTを安定化させ,グルタチオンを増加させることにより活性酸素種の蓄積を防いでいることが明らかにされている10).
CD44v9陽性がん幹細胞ではm1型VacAによるグルタチオンの減少に阻害を示し,CagA分解性オートファジーが起こらずCagAが蓄積するのではないかと仮説をたてた.そこで,CD44陰性のMKN28細胞に通常型のCD44sおよびCD44v9をそれぞれ強制発現させたのち,ピロリ菌に感染した細胞におけるグルタチオンのレベルを検討した.CD44sを強制発現させた細胞とは対照的に,CD44v9を強制発現させた細胞ではピロリ菌に感染してもグルタチオンは減少しなかった.その結果,AktのSer473のリン酸化,Mdm2のリン酸化,p53の減少は誘導されず,オートファジーの誘導は抑制されCagAの蓄積が認められた.また,CD44v9を強制発現させたMKN28細胞にピロリ菌を感染させxCTの阻害剤であるスルファサラジンを処理すると,Aktのリン酸化およびMdm2のリン酸化が誘導されp53は減少し,CagA分解性オートファジーの誘導とともにCagAの分解が生じた.これらの結果から,CD44v9を発現する細胞におけるCagAの蓄積は,CD44v9によるxCTの安定化を介したグルタチオンの増加に依存していることが示された(図2).
おわりに
宿主細胞に移行したCagAはSHP2およびPAR1との相互作用を介し異常な細胞増殖を惹起し上皮細胞の極性を破壊する.そのため,宿主細胞におけるCagAの過剰な安定は上皮細胞の脱落を容易に誘発し,ピロリ菌にとり,定着した足場の喪失につながる.そこで,ピロリ菌は胃における長期的な生存戦略のひとつとして,VacAによるオートファジーの誘導を介しCagAを負に制御する能力を獲得したとも考えられる.一方で,CD44v9陽性がん幹細胞はVacAによるCagA分解性オートファジーの誘導に阻害を示すことによりCagAを蓄積する.したがって,CD44v9陽性がん幹細胞の存在はピロリ菌に感染した際の胃がんの発がんに強く関与し,また,胃がんの発がんリスクを亢進させる要因になると考えられる.この研究により,ピロリ菌とCD44v9陽性がん幹細胞との遭遇が,ヒトにとりピロリ菌に感染した際の胃がんの発がんリスクを規定する要因となることが示され,これまでの機序では説明できなかった,ピロリ菌の感染者すべてが胃がんを発症するのではないという事実の一部を説明することになった.今後の胃がんの予防および治療の標的として,ピロリ菌の感染という根本的な標的はいうまでもなく,長期の炎症を背景に出現すると考えられるCD44v9陽性がん幹細胞についてもその重要性を提示することができたわけである.
文 献
- Hatakeyama, M.: Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat. Rev. Cancer, 4, 688-694 (2004)[PubMed]
- Higashi, H., Tsutsumi, R., Muto, S. et al.: SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science, 295, 683-686 (2002)[PubMed]
- Ohnishi, N., Yuasa, H., Tanaka, S. et al.: Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc. Natl. Acad. Sci. USA, 105, 1003-1008 (2008)[PubMed]
- Ishikawa, S., Ohta, T. & Hatakeyama, M.: Stability of Helicobacter pylori CagA oncoprotein in human gastric epithelial cells. FEBS Lett., 583, 2414-2418 (2009)[PubMed]
- Terebiznik, M. R., Raju, D., Vazquez, C. L. et al.: Effect of Helicobacter pylori's vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy, 5, 370-379 (2009)[PubMed]
- Raju, D., Hussey, S., Ang, M. et al.: Vacuolating cytotoxin and variants in Atg16L1 that disrupt autophagy promote Helicobacter pylori infection in humans. Gastroenterology, 142, 1160-1171 (2012)[PubMed]
- Yahiro, K., Satoh, M., Nakano, M. et al.: Low-density lipoprotein receptor-related protein-1 (LRP1) mediates autophagy and apoptosis caused by Helicobacter pylori VacA. J. Biol. Chem., 287, 31104-31115 (2012)[PubMed]
- Gangwer, K. A., Mushrush, D. J., Stauff, D. L. et al.: Crystal structure of the Helicobacter pylori vacuolating toxin p55 domain. Proc. Natl. Acad. Sci. USA, 104, 16293-16298 (2007)[PubMed]
- Tasdemir, E., Maiuri, M. C., Galluzzi, L. et al.: Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol., 10, 676-687 (2008)[PubMed]
- Ishimoto, T., Nagano, O., Yae, T. et al.: CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc- and thereby promotes tumor growth. Cancer Cell, 19, 387-400 (2011)[PubMed]
著者プロフィール
略歴:2007年 東北薬科大学大学院薬学研究科博士課程 修了,同年 慶應義塾大学医学部 共同研究員を経て,同年より同 特任助教.
研究テーマ:ピロリ菌の感染の分子機構と発がん.
関心事:細菌と宿主との攻防戦と,その結果,生じる病態の解明.
鈴木 秀和(Hidekazu Suzuki)
慶應義塾大学医学部 准教授.
© 2012 津川 仁・鈴木秀和 Licensed under CC 表示 2.1 日本
