ナラトリプタンはCGRP1の発現抑制を介し球脊髄性筋萎縮症を抑止する
南山 誠・勝野雅央・祖父江 元
(名古屋大学大学院医学系研究科 神経内科学)
email:勝野雅央,祖父江 元
DOI: 10.7875/first.author.2012.128
Naratriptan mitigates CGRP1-associated motor neuron degeneration caused by an expanded polyglutamine repeat tract.
Makoto Minamiyama, Masahisa Katsuno, Hiroaki Adachi, Hideki Doi, Naohide Kondo, Madoka Iida, Shinsuke Ishigaki, Yusuke Fujioka, Shinjiro Matsumoto, Yu Miyazaki, Fumiaki Tanaka, Hiroki Kurihara, Gen Sobue
Nature Medicine, 18, 1531-1538 (2012)
球脊髄性筋萎縮症はアンドロゲン受容体遺伝子のもつCAGくり返し配列の異常な延長を原因とする神経変性疾患である.今回,筆者らは,球脊髄性筋萎縮症モデルマウスの脊髄から抽出したmRNAに対しマイクロアレイ解析を行い,発現の有意に上昇しているタンパク質としてCGRP1を同定した.神経系の培養細胞においてこのCGRP1を過剰発現したところ,JNKシグナルの活性化を介し細胞死が誘導された.CGRP1のノックアウトにより,球脊髄性筋萎縮症モデルマウスの運動ニューロンにおいてJNK経路が抑制されたとともに,運動機能や寿命に有意な改善が認められた.セロトニン受容体アゴニストであるナラトリプタンは神経系の培養細胞においてCGRP1の発現量を低下させ,変異アンドロゲン受容体による細胞障害を軽減した.ナラトリプタンを球脊髄性筋萎縮症モデルマウスに経口投与したところ,運動ニューロンにおいてCGRP1の発現量は減少し,JNKシグナルが抑制され運動機能や寿命が有意に改善した.
アルツハイマー病,パーキンソン病,運動ニューロン病などに代表される神経変性疾患は,特定の神経が変性し脱落することにより進行性かつ難治性の認知機能および運動機能の障害を呈する.種々の神経変性疾患に共通した病理学的な特徴は変性したニューロンの内外への異常なタンパク質の沈着であり,異常なタンパク質が生理的な防御機構を凌駕し蓄積することが神経変性の中心的な病態であると考えられている.しかし,ニューロンやその周囲に蓄積した異常なタンパク質がどのようにニューロンの機能障害や細胞死を誘導するのかについては不明な点が多く,いまもって病態そのものを抑止する根本的な治療法は確立されていない.今回,筆者らは,運動ニューロンの変性を呈する神経変性疾患のひとつである球脊髄性筋萎縮症(spinal and bulbar muscular atrophy:SBMA)を対象とし,神経変性に関与する細胞におけるシグナル異常を標的とした治療法の開発を目的として,球脊髄性筋萎縮症のモデルマウスを用いた検討を行った.
球脊髄性筋萎縮症は成人に発症する運動ニューロン疾患であり,脊髄および脳幹の下位運動ニューロンにおける選択的な変性と脱落を呈する神経変性疾患である.1897年,わが国において進行性球麻痺の兄弟例が記録されたのが球脊髄性筋萎縮症における世界初の報告であり,1968年に,11例の臨床像および病理像がまとめて発表されたことにより単一の疾患として認められるようになった1).球脊髄性筋萎縮症の特徴のひとつは男性のみに発症することであり,有病率は人口10万人あたり1~2人と推定されている.主症状は四肢の筋力低下および筋萎縮と球麻痺(咽頭筋の筋力低下)であり,四肢の運動障害は近位部においてとくに強くみられ,動揺性歩行や起立困難となる.はじめて筋力低下を自覚する時期は30~60歳だが,手指の振戦や有痛性の筋痙攣,女性化乳房などのアンドロゲン不応症状がしばしば筋力低下に先行する.根本的な治療法は存在せず,対症療法としてテストステロンが使用されることがあったが有効性は認められていない.進行性の経過をたどり,呼吸器への感染などが死因になることが多いと報告されている.
球脊髄性筋萎縮症の原因はアンドロゲン受容体遺伝子の第1エクソンにおけるCAGくり返し配列(CAGリピート)の異常な延長である2).アンドロゲン受容体遺伝子のもつCAGくり返し配列の数は正常においては9~36であるが,球脊髄性筋萎縮症の患者すべてにおいては38以上に延長しており,CAGくり返し配列の数が大きいほど発症年齢は若年となることが知られている.同様にCAGくり返し配列の異常な延長を原因とする疾患としてハンチントン病や脊髄小脳失調症が知られており,これらポリグルタミン病とよばれる疾患では,異常に伸長したポリグルタミン鎖をもつ変異タンパク質がニューロンに蓄積することが病態の根幹と考えられている3).病理学的には,脊髄前角や脳幹の運動ニューロンの変性と脱落が認められ,残存する運動ニューロンの核には変異アンドロゲン受容体の異常な集積がみられる.
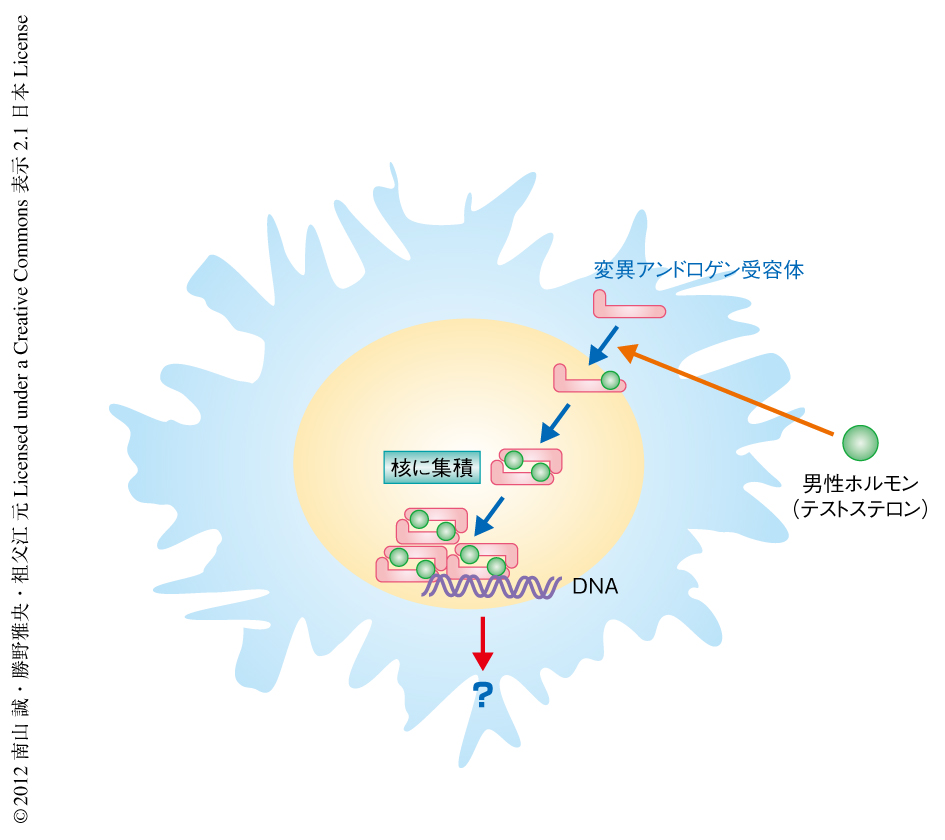
球脊髄性筋萎縮症の原因タンパク質となるアンドロゲン受容体は,通常は熱ショックタンパク質などと複合体を形成し不活性化された状態で細胞質に存在するが,リガンドである男性ホルモン(テストステロン)が存在するとこれらのタンパク質とは離れ核へと移行する.筆者らは,球脊髄性筋萎縮症の動物モデルとして,CAGくり返し配列の数が97に異常延長したヒトアンドロゲン受容体遺伝子を発現するトランスジェニックマウスを作製した.このモデルマウスでは,球脊髄性筋萎縮症の患者と同様の進行性の筋力低下や神経原性の筋萎縮が認められ,これらの所見はメスに比べオスにおいてより強く観察される4).症状の重症なオスの球脊髄性筋萎縮症モデルマウスに去勢術を行うと,変異アンドロゲン受容体の核における集積は著明に抑制され,筋力低下などの症状や寿命も著しく改善する.逆に,メスの球脊髄性筋萎縮症モデルマウスにテストステロンを投与すると,オスと同様の顕著な筋力低下および筋萎縮が生じ,病理学的にも変異アンドロゲン受容体の核における集積が増悪する.これらの結果は,変異アンドロゲン受容体がテストステロンの濃度に依存してニューロンの核に集積することが球脊髄性筋萎縮症の病態の根幹であることを示唆しており,同様の解析結果は,ほかの動物モデルや,テストステロンの分泌を抑制する効果をもつleuprorelinを用いた検討においても報告されている5).核に集積した変異アンドロゲン受容体は転写の阻害や軸索輸送の阻害などをもたらし,これらの異常が運動ニューロンの変性に関与すると考えられているが,その詳細については不明な点が多い6)(図1).
球脊髄性筋萎縮症における運動ニューロンの変性の分子機序を明らかにするため,CAGくり返し配列の数が97に異常延長したヒトアンドロゲン受容体を発現する球脊髄性筋萎縮症モデルマウスの脊髄から抽出したmRNAに対しマイクロアレイ解析を行った.発症前(7~9週齢),発症早期(10~12週齢),進行期(13~15週齢)のそれぞれについて,球脊髄性筋萎縮症モデルマウスと2種類の対照マウス(正常な長さのCAGくり返し配列をもつヒトアンドロゲン受容体を発現するトランスジェニックマウス,および,野生型マウス)とのあいだで遺伝子の発現を比較したところ,対照となるマウスと比較し,球脊髄性筋萎縮症モデルマウスにおいて運動障害の発症するまえから転写の有意に変化している13の遺伝子が同定された.これらのうち,多機能神経ペプチドをコードするCGRP1(calcitonin gene-related peptide 1,カルシトニン遺伝子関連ペプチド1)遺伝子の発現がもっとも著しく変化(上昇)しており,中枢神経系ではとくに運動ニューロンにおいて発現が高く,球脊髄性筋萎縮症モデルマウスと対照となるマウスとのあいだの発現の差は病態の進行とともに拡大することを見い出した.CGRP1遺伝子は球脊髄性筋萎縮症モデルマウスのみならず球脊髄性筋萎縮症の患者の運動ニューロンにおいても発現量が増加しており,球脊髄性筋萎縮症モデルマウスでは去勢による病態の改善とともに運動ニューロンにおける発現量が低下した.
球脊髄性筋萎縮症におけるCGRP1の発現亢進の機序を明らかにするため,ヒトの神経芽細胞腫に由来する培養細胞であるSH-SY5Y細胞においてCAGくり返し配列の数が97に異常延長した変異ヒトアンドロゲン受容体を強制発現させたところ,CGRP1遺伝子のプロモーター活性が上昇し,そのmRNAおよびタンパク質の発現量が増加した.この結果から,変異アンドロゲン受容体はCGRP1遺伝子の転写を亢進させることによりその発現量を増加させると考えられた.CGRP1の発現量の増加はニューロンにとり有害なのかどうかを調べたところ,SH-SY5Y細胞におけるCGRP1の過剰発現,あるいは,SH-SY5Y細胞の培養液へのCGRP1の添加により細胞死が誘導され,CGRP1のノックダウン,あるいは,ペプチドを用いたCGRP1の薬物学的な阻害により細胞死は抑制された.その分子機序として,CGRP1がc-Junのリン酸化を介しJNKシグナルを活性化すること,および,変異アンドロゲン受容体のもつ細胞毒性がJNK経路の阻害剤により抑制されることも明らかになった.球脊髄性筋萎縮症モデルマウスとCGRP1ノックアウトマウスとを交配し症状や病理学的な所見の変化を解析したところ,CGRP1のノックアウトにより運動機能や寿命に有意な改善が認められた.また,神経変性にともなうアストロサイトの増生の指標であるGFAP(glial fibrillary acidic protein,グリア線維性酸性タンパク質)の発現量は減少し,運動ニューロンの機能の指標であるコリンアセチルトランスフェラーゼの発現量は増加した.骨格筋では運動ニューロンの変性の結果として生じる筋萎縮が改善した.JNK活性の指標であるc-Junのリン酸化は対照マウスに比べ球脊髄性筋萎縮症モデルマウスの運動ニューロンにおいて活性化されていたが,これもCGRP1のノックアウトにより有意に改善した.以上より,CGRP1によるJNKの異常な活性化が球脊髄性筋萎縮症における神経変性の機序として重要であることが示唆された.
CGRP1は片頭痛の病態に関連するタンパク質としても知られている7).そこで,CGRP1-JNK経路をターゲットとした治療法の開発を目的として,CGRP1の発現を抑制する低分子化合物をスクリーニングしたところ,片頭痛の治療薬として使用されているナラトリプタンなどのセロトニン受容体アゴニストがCGRP1の発現量を低下させることが明らかになった.そこで,CAGくり返し配列の数が97に異常延長した変異アンドロゲン受容体を過剰発現させたSH-SY5Y細胞にナラトリプタンを投与したところ,細胞増殖試薬WST-1を用い測定された細胞活性は改善し,細胞死は抑制されることが示された.さらに,ナラトリプタンによるCGRP1の発現抑制の分子機構として,ナラトリプタンがMAPキナーゼの抑制タンパク質のひとつであるDUSP1(dual specificity protein phosphatase 1)の発現を誘導すること,および,DUSP1のノックダウンや薬物学的な阻害によりナラトリプタンのもつニューロンの保護効果が減弱することが明らかになった.ナラトリプタンを球脊髄性筋萎縮症モデルマウスに経口投与したところ,運動機能は改善し生存期間は約1.5倍に延長した.病理学的にも,脊髄におけるGFAPの発現量は減少し,コリンアセチルトランスフェラーゼの発現量が増加するとともに,JNK活性の指標であるc-Junのリン酸化も有意に改善した.骨格筋においては,ナラトリプタンにより運動ニューロンの変性の結果として生じる筋萎縮が改善した.
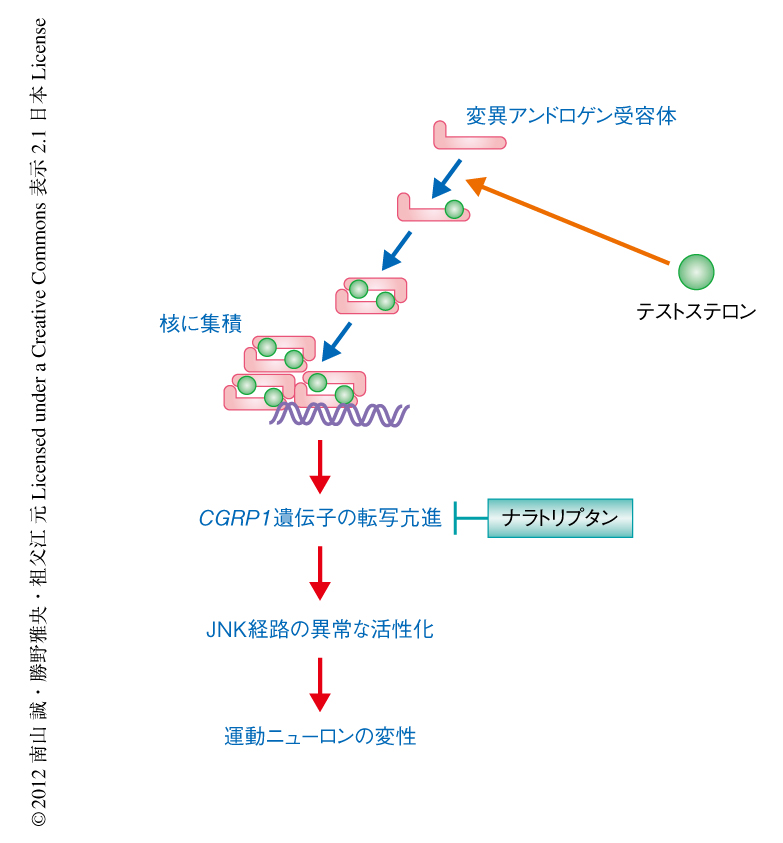
ナラトリプタンによるCGRP1-JNK経路の抑制はポリグルタミン鎖に関連した運動ニューロンの変性を抑制する治療法になりうると考えられる(図2).現在,球脊髄性筋萎縮症に対しテストステロンの抑制による治療法の開発が進められており8,9),今後,ナラトリプタンの効果についても臨床試験により検証することを検討する予定である.また,CGRP1は別の運動ニューロン疾患である筋萎縮性側索硬化症の病態に関与するとの報告もあり10),球脊髄性筋萎縮症の病態との共通性について,今後,さらなる解析が必要である.
略歴:2005年 名古屋大学大学院医学系研究科 修了,同年 同 客員研究員を経て,2012年より国立長寿医療研究センター研究所 室長.
研究テーマ:神経変性疾患.
関心事:神経変性疾患の治療法の開発,遺伝カウンセリング.
勝野 雅央(Masahisa Katsuno)
略歴:2003年 名古屋大学大学院医学系研究科 修了,2004年 長寿科学振興財団 リサーチレジデント,2006年 名古屋大学高等研究院 特任講師を経て,2011年より名古屋大学大学院医学系 特任准教授.
研究テーマ:神経変性疾患,分子生物学,臨床試験.
関心事:神経変性疾患に対するトランスレーショナルリサーチ.
祖父江 元(Gen Sobue)
名古屋大学大学院医学系研究科 教授.
© 2012 南山 誠・勝野雅央・祖父江 元 Licensed under CC 表示 2.1 日本
(名古屋大学大学院医学系研究科 神経内科学)
email:勝野雅央,祖父江 元
DOI: 10.7875/first.author.2012.128
Naratriptan mitigates CGRP1-associated motor neuron degeneration caused by an expanded polyglutamine repeat tract.
Makoto Minamiyama, Masahisa Katsuno, Hiroaki Adachi, Hideki Doi, Naohide Kondo, Madoka Iida, Shinsuke Ishigaki, Yusuke Fujioka, Shinjiro Matsumoto, Yu Miyazaki, Fumiaki Tanaka, Hiroki Kurihara, Gen Sobue
Nature Medicine, 18, 1531-1538 (2012)
要 約
球脊髄性筋萎縮症はアンドロゲン受容体遺伝子のもつCAGくり返し配列の異常な延長を原因とする神経変性疾患である.今回,筆者らは,球脊髄性筋萎縮症モデルマウスの脊髄から抽出したmRNAに対しマイクロアレイ解析を行い,発現の有意に上昇しているタンパク質としてCGRP1を同定した.神経系の培養細胞においてこのCGRP1を過剰発現したところ,JNKシグナルの活性化を介し細胞死が誘導された.CGRP1のノックアウトにより,球脊髄性筋萎縮症モデルマウスの運動ニューロンにおいてJNK経路が抑制されたとともに,運動機能や寿命に有意な改善が認められた.セロトニン受容体アゴニストであるナラトリプタンは神経系の培養細胞においてCGRP1の発現量を低下させ,変異アンドロゲン受容体による細胞障害を軽減した.ナラトリプタンを球脊髄性筋萎縮症モデルマウスに経口投与したところ,運動ニューロンにおいてCGRP1の発現量は減少し,JNKシグナルが抑制され運動機能や寿命が有意に改善した.
はじめに
アルツハイマー病,パーキンソン病,運動ニューロン病などに代表される神経変性疾患は,特定の神経が変性し脱落することにより進行性かつ難治性の認知機能および運動機能の障害を呈する.種々の神経変性疾患に共通した病理学的な特徴は変性したニューロンの内外への異常なタンパク質の沈着であり,異常なタンパク質が生理的な防御機構を凌駕し蓄積することが神経変性の中心的な病態であると考えられている.しかし,ニューロンやその周囲に蓄積した異常なタンパク質がどのようにニューロンの機能障害や細胞死を誘導するのかについては不明な点が多く,いまもって病態そのものを抑止する根本的な治療法は確立されていない.今回,筆者らは,運動ニューロンの変性を呈する神経変性疾患のひとつである球脊髄性筋萎縮症(spinal and bulbar muscular atrophy:SBMA)を対象とし,神経変性に関与する細胞におけるシグナル異常を標的とした治療法の開発を目的として,球脊髄性筋萎縮症のモデルマウスを用いた検討を行った.
球脊髄性筋萎縮症は成人に発症する運動ニューロン疾患であり,脊髄および脳幹の下位運動ニューロンにおける選択的な変性と脱落を呈する神経変性疾患である.1897年,わが国において進行性球麻痺の兄弟例が記録されたのが球脊髄性筋萎縮症における世界初の報告であり,1968年に,11例の臨床像および病理像がまとめて発表されたことにより単一の疾患として認められるようになった1).球脊髄性筋萎縮症の特徴のひとつは男性のみに発症することであり,有病率は人口10万人あたり1~2人と推定されている.主症状は四肢の筋力低下および筋萎縮と球麻痺(咽頭筋の筋力低下)であり,四肢の運動障害は近位部においてとくに強くみられ,動揺性歩行や起立困難となる.はじめて筋力低下を自覚する時期は30~60歳だが,手指の振戦や有痛性の筋痙攣,女性化乳房などのアンドロゲン不応症状がしばしば筋力低下に先行する.根本的な治療法は存在せず,対症療法としてテストステロンが使用されることがあったが有効性は認められていない.進行性の経過をたどり,呼吸器への感染などが死因になることが多いと報告されている.
球脊髄性筋萎縮症の原因はアンドロゲン受容体遺伝子の第1エクソンにおけるCAGくり返し配列(CAGリピート)の異常な延長である2).アンドロゲン受容体遺伝子のもつCAGくり返し配列の数は正常においては9~36であるが,球脊髄性筋萎縮症の患者すべてにおいては38以上に延長しており,CAGくり返し配列の数が大きいほど発症年齢は若年となることが知られている.同様にCAGくり返し配列の異常な延長を原因とする疾患としてハンチントン病や脊髄小脳失調症が知られており,これらポリグルタミン病とよばれる疾患では,異常に伸長したポリグルタミン鎖をもつ変異タンパク質がニューロンに蓄積することが病態の根幹と考えられている3).病理学的には,脊髄前角や脳幹の運動ニューロンの変性と脱落が認められ,残存する運動ニューロンの核には変異アンドロゲン受容体の異常な集積がみられる.
球脊髄性筋萎縮症の原因タンパク質となるアンドロゲン受容体は,通常は熱ショックタンパク質などと複合体を形成し不活性化された状態で細胞質に存在するが,リガンドである男性ホルモン(テストステロン)が存在するとこれらのタンパク質とは離れ核へと移行する.筆者らは,球脊髄性筋萎縮症の動物モデルとして,CAGくり返し配列の数が97に異常延長したヒトアンドロゲン受容体遺伝子を発現するトランスジェニックマウスを作製した.このモデルマウスでは,球脊髄性筋萎縮症の患者と同様の進行性の筋力低下や神経原性の筋萎縮が認められ,これらの所見はメスに比べオスにおいてより強く観察される4).症状の重症なオスの球脊髄性筋萎縮症モデルマウスに去勢術を行うと,変異アンドロゲン受容体の核における集積は著明に抑制され,筋力低下などの症状や寿命も著しく改善する.逆に,メスの球脊髄性筋萎縮症モデルマウスにテストステロンを投与すると,オスと同様の顕著な筋力低下および筋萎縮が生じ,病理学的にも変異アンドロゲン受容体の核における集積が増悪する.これらの結果は,変異アンドロゲン受容体がテストステロンの濃度に依存してニューロンの核に集積することが球脊髄性筋萎縮症の病態の根幹であることを示唆しており,同様の解析結果は,ほかの動物モデルや,テストステロンの分泌を抑制する効果をもつleuprorelinを用いた検討においても報告されている5).核に集積した変異アンドロゲン受容体は転写の阻害や軸索輸送の阻害などをもたらし,これらの異常が運動ニューロンの変性に関与すると考えられているが,その詳細については不明な点が多い6)(図1).
1.球脊髄性筋萎縮症におけるCGRP1-JNK経路の異常
球脊髄性筋萎縮症における運動ニューロンの変性の分子機序を明らかにするため,CAGくり返し配列の数が97に異常延長したヒトアンドロゲン受容体を発現する球脊髄性筋萎縮症モデルマウスの脊髄から抽出したmRNAに対しマイクロアレイ解析を行った.発症前(7~9週齢),発症早期(10~12週齢),進行期(13~15週齢)のそれぞれについて,球脊髄性筋萎縮症モデルマウスと2種類の対照マウス(正常な長さのCAGくり返し配列をもつヒトアンドロゲン受容体を発現するトランスジェニックマウス,および,野生型マウス)とのあいだで遺伝子の発現を比較したところ,対照となるマウスと比較し,球脊髄性筋萎縮症モデルマウスにおいて運動障害の発症するまえから転写の有意に変化している13の遺伝子が同定された.これらのうち,多機能神経ペプチドをコードするCGRP1(calcitonin gene-related peptide 1,カルシトニン遺伝子関連ペプチド1)遺伝子の発現がもっとも著しく変化(上昇)しており,中枢神経系ではとくに運動ニューロンにおいて発現が高く,球脊髄性筋萎縮症モデルマウスと対照となるマウスとのあいだの発現の差は病態の進行とともに拡大することを見い出した.CGRP1遺伝子は球脊髄性筋萎縮症モデルマウスのみならず球脊髄性筋萎縮症の患者の運動ニューロンにおいても発現量が増加しており,球脊髄性筋萎縮症モデルマウスでは去勢による病態の改善とともに運動ニューロンにおける発現量が低下した.
球脊髄性筋萎縮症におけるCGRP1の発現亢進の機序を明らかにするため,ヒトの神経芽細胞腫に由来する培養細胞であるSH-SY5Y細胞においてCAGくり返し配列の数が97に異常延長した変異ヒトアンドロゲン受容体を強制発現させたところ,CGRP1遺伝子のプロモーター活性が上昇し,そのmRNAおよびタンパク質の発現量が増加した.この結果から,変異アンドロゲン受容体はCGRP1遺伝子の転写を亢進させることによりその発現量を増加させると考えられた.CGRP1の発現量の増加はニューロンにとり有害なのかどうかを調べたところ,SH-SY5Y細胞におけるCGRP1の過剰発現,あるいは,SH-SY5Y細胞の培養液へのCGRP1の添加により細胞死が誘導され,CGRP1のノックダウン,あるいは,ペプチドを用いたCGRP1の薬物学的な阻害により細胞死は抑制された.その分子機序として,CGRP1がc-Junのリン酸化を介しJNKシグナルを活性化すること,および,変異アンドロゲン受容体のもつ細胞毒性がJNK経路の阻害剤により抑制されることも明らかになった.球脊髄性筋萎縮症モデルマウスとCGRP1ノックアウトマウスとを交配し症状や病理学的な所見の変化を解析したところ,CGRP1のノックアウトにより運動機能や寿命に有意な改善が認められた.また,神経変性にともなうアストロサイトの増生の指標であるGFAP(glial fibrillary acidic protein,グリア線維性酸性タンパク質)の発現量は減少し,運動ニューロンの機能の指標であるコリンアセチルトランスフェラーゼの発現量は増加した.骨格筋では運動ニューロンの変性の結果として生じる筋萎縮が改善した.JNK活性の指標であるc-Junのリン酸化は対照マウスに比べ球脊髄性筋萎縮症モデルマウスの運動ニューロンにおいて活性化されていたが,これもCGRP1のノックアウトにより有意に改善した.以上より,CGRP1によるJNKの異常な活性化が球脊髄性筋萎縮症における神経変性の機序として重要であることが示唆された.
2.CGRP1を標的とした球脊髄性筋萎縮症の治療法の開発
CGRP1は片頭痛の病態に関連するタンパク質としても知られている7).そこで,CGRP1-JNK経路をターゲットとした治療法の開発を目的として,CGRP1の発現を抑制する低分子化合物をスクリーニングしたところ,片頭痛の治療薬として使用されているナラトリプタンなどのセロトニン受容体アゴニストがCGRP1の発現量を低下させることが明らかになった.そこで,CAGくり返し配列の数が97に異常延長した変異アンドロゲン受容体を過剰発現させたSH-SY5Y細胞にナラトリプタンを投与したところ,細胞増殖試薬WST-1を用い測定された細胞活性は改善し,細胞死は抑制されることが示された.さらに,ナラトリプタンによるCGRP1の発現抑制の分子機構として,ナラトリプタンがMAPキナーゼの抑制タンパク質のひとつであるDUSP1(dual specificity protein phosphatase 1)の発現を誘導すること,および,DUSP1のノックダウンや薬物学的な阻害によりナラトリプタンのもつニューロンの保護効果が減弱することが明らかになった.ナラトリプタンを球脊髄性筋萎縮症モデルマウスに経口投与したところ,運動機能は改善し生存期間は約1.5倍に延長した.病理学的にも,脊髄におけるGFAPの発現量は減少し,コリンアセチルトランスフェラーゼの発現量が増加するとともに,JNK活性の指標であるc-Junのリン酸化も有意に改善した.骨格筋においては,ナラトリプタンにより運動ニューロンの変性の結果として生じる筋萎縮が改善した.
おわりに
ナラトリプタンによるCGRP1-JNK経路の抑制はポリグルタミン鎖に関連した運動ニューロンの変性を抑制する治療法になりうると考えられる(図2).現在,球脊髄性筋萎縮症に対しテストステロンの抑制による治療法の開発が進められており8,9),今後,ナラトリプタンの効果についても臨床試験により検証することを検討する予定である.また,CGRP1は別の運動ニューロン疾患である筋萎縮性側索硬化症の病態に関与するとの報告もあり10),球脊髄性筋萎縮症の病態との共通性について,今後,さらなる解析が必要である.
文 献
- Katsuno, M., Adachi, H., Waza, M. et al.: Pathogenesis, animal models and therapeutics in spinal and bulbar muscular atrophy (SBMA). Exp. Neurol., 200, 8-18 (2006)[PubMed]
- La Spada, A. R., Wilson, E. M., Lubahn, D. B. et al.: Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature, 352, 77-79 (1991)[PubMed]
- Ross, C. A. & Tabrizi, S. J.: Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol., 10, 83-98 (2011)[PubMed]
- Katsuno, M., Adachi, H., Kume, A. et al.: Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron, 35, 843-854 (2002)[PubMed]
- Katsuno, M., Adachi, H., Doyu, M. et al.: Leuprorelin rescues polyglutamine-dependent phenotypes in a transgenic mouse model of spinal and bulbar muscular atrophy. Nat. Med., 9, 768-773 (2003)[PubMed]
- Minamiyama, M., Katsuno, M., Adachi, H. et al.: Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum. Mol. Genet., 13, 1183-1192 (2004)[PubMed]
- Ho, T. W., Edvinsson, L., Goadsby, P. J.: CGRP and its receptors provide new insights into migraine pathophysiology. Nat. Rev. Neurol., 6, 573-582 (2010)[PubMed]
- Katsuno, M., Banno, H., Suzuki, K. et al.: Efficacy and safety of leuprorelin in patients with spinal and bulbar muscular atrophy (JASMITT study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol., 9, 875-884 (2010)[PubMed]
- Fernandez-Rhodes, L. E., Kokkinis, A. D., White, M. J. et al.: Efficacy and safety of dutasteride in patients with spinal and bulbar muscular atrophy: a randomised placebo-controlled trial. Lancet Neurol., 10, 140-147 (2011)[PubMed]
- Ringer, C., Weihe, E., Schutz, B.: Calcitonin gene-related peptide expression levels predict motor neuron vulnerability in the superoxide dismutase 1-G93A mouse model of amyotrophic lateral sclerosis. Neurobiol., Dis. 45, 547-554 (2011)[PubMed]
著者プロフィール
略歴:2005年 名古屋大学大学院医学系研究科 修了,同年 同 客員研究員を経て,2012年より国立長寿医療研究センター研究所 室長.
研究テーマ:神経変性疾患.
関心事:神経変性疾患の治療法の開発,遺伝カウンセリング.
勝野 雅央(Masahisa Katsuno)
略歴:2003年 名古屋大学大学院医学系研究科 修了,2004年 長寿科学振興財団 リサーチレジデント,2006年 名古屋大学高等研究院 特任講師を経て,2011年より名古屋大学大学院医学系 特任准教授.
研究テーマ:神経変性疾患,分子生物学,臨床試験.
関心事:神経変性疾患に対するトランスレーショナルリサーチ.
祖父江 元(Gen Sobue)
名古屋大学大学院医学系研究科 教授.
© 2012 南山 誠・勝野雅央・祖父江 元 Licensed under CC 表示 2.1 日本
