病的血管新生はATMによる酸化ストレス調節に依存する
奥野祐次・久保田義顕
(慶應義塾大学医学部 総合医科研究センター咸臨丸プロジェクト)
email:久保田義顕
DOI: 10.7875/first.author.2012.090
Pathological neoangiogenesis depends on oxidative stress regulation by ATM.
Yuji Okuno, Ayako Nakamura-Ishizu, Kinya Otsu, Toshio Suda, Yoshiaki Kubota
Nature Medicine, 18, 1208-1212 (2012)
AtmキナーゼはDNA損傷応答におけるマスター制御タンパク質であり,細胞の老化や腫瘍の形成に対する障壁として機能することが広く知られている.筆者らは,Atmキナーゼが活性酸素種の蓄積に応答し未成熟な血管において特異的に活性化されることを見い出した.マウスにおいて全身あるいは血管内皮に特異的にAtm遺伝子を欠損させると網膜における病的な血管新生が阻害された.この阻害はDNA損傷応答の異常ではなく,活性酸素種の増加およびp38αの過剰な活性化によりひき起こされていた.また,Atm遺伝子の欠損は腫瘍における血管新生の著明な減少,および,VEGFシグナルの阻害による抗血管新生作用の亢進もひき起こした.これらの結果は,病的な血管新生にはAtmキナーゼを介する酸化ストレスの制御機構が必要であること,また,活性酸素種の過剰な産生を促進する薬剤が血管新生病の治療に効果をもつ可能性を示唆した.
生体のあらゆる組織がその恒常性を維持するためには,血管網から過不足なく酸素および栄養を供給されることが必須である.また,この組織への酸素供給に最適な血管のパターニングを規定する分子機構は,発生期における血管新生だけではなく,血管の過剰な増殖に端を発する血管新生病の病態の進行においても重要である.とくに,糖尿病性網膜症,加齢性黄斑変性症など,成人において失明の大きな原因のひとつとなる網膜血管新生病において,その病態の鍵となっている.さらには,がんの増大および進行においても,既存の血管からの血管新生がその病態に深くかかわっている.現在,臨床の場で用いられている抗がん剤の多くは腫瘍細胞の旺盛に増殖するという特徴を標的としたものであり,DNAやタンパク質の合成などの細胞分裂の過程を傷害するため,制がん効果の高いものほど正常組織,とくに,細胞のターンオーバーの速い造血細胞,消化管,皮膚などへのダメージも大きく,造血や消化管の障害など副作用が大きい.一方,腫瘍血管を標的とした“抗腫瘍血管新生療法”は,理論上,従来の抗がん剤に比べ正常な組織へのダメージは少なく,がんの内科的な治療におけるひとつのオプションとして確立されつつある.この抗腫瘍血管新生療法として中心的な戦略が,VEGF(vascular endothelial growth factor,血管内皮増殖因子)シグナルの阻害である.VEGFの中和抗体であるBevacizumabは,2004年の米国食品医薬品局,2007年の厚生労働省の認可ののち,臨床の場で広く用いられ,大腸がん,肺がん,腎がんなど多くのがん,とくに,ほかの内科的な治療に抵抗性の症例において有意な抗腫瘍効果をもつことが判明している1).ところが,一部のがんはこのVEGF阻害剤に対し耐性あるいは抵抗性を示し,とくに,VEGF阻害剤の投与を中断したのちに急激な血管の再成長をともなう腫瘍の増大をひき起こすことが判明しつつある2).さらには,VEGF阻害剤は腫瘍血管のみならず健常な血管をも傷害し,脳血管の障害や消化管の出血など,しばしば致命的な副作用を及ぼすことが報告されている3).これらを背景に,VEGFの阻害に代わる,または,併用しうる新たな抗血管新生療法の分子標的が世界的に探索されている.なかでも,腫瘍や網膜血管新生病における病的な血管のみに機能するタンパク質,つまり,病的な血管新生に選択的な治療標的が広く探索されている.
Atm(Ataxia telangiectasia mutated)遺伝子はヒトの毛細血管拡張性肉芽腫の原因遺伝子として知られている.Atm遺伝子のコードするAtmキナーゼはDNA損傷応答における中心的なタンパク質としてその機能はよく知られており,毛細血管拡張性肉芽腫の患者には高率(約30%)にがんが発生する4).しかしながら,毛細血管拡張性肉芽腫の患者に発生するがんは,ほとんどがリンパ腫や白血病などの腫瘍微小環境(がんニッチ)への依存性の低い造血系の腫瘍であり,固形腫瘍の発生はまれである5).これらの知見から,この研究は,Atmキナーゼの機能喪失による血管新生の異常がみかけ上,固形腫瘍の発生を覆い隠しているのではないかという仮説のもと,血管におけるAtmキナーゼの活性化および機能を解析した.
平常時および病態における血管内皮細胞でのAtmキナーゼの活性化を検出するため,正常に発生したマウスの網膜,および,病的な血管新生のモデルである虚血性網膜症モデル6) を施したマウスの網膜に対し,Atmキナーゼ活性化の指標となる抗リン酸化Atmキナーゼ抗体による免疫染色を行った.その結果,リン酸化Atmキナーゼは正常な血管においてはほとんど検出されないものの,病的な血管新生において強く検出された.また,この病的な血管新生の生じた部位では血管内皮がジヒドロエチジウムにより強く染色され,活性酸素種の蓄積が示唆された.虚血性網膜症モデルを施したマウスを抗酸化剤であるN-アセチル-L-システインにより処理したところ,病的な血管新生の生じた部位におけるリン酸化Atmキナーゼの免疫染色の強度は,正常に発生した網膜の血管のレベルまで下がった.以上の結果から,Atmキナーゼはマウスの網膜において酸化ストレスに応じ病的な血管に特異的に活性化されていることが示された.
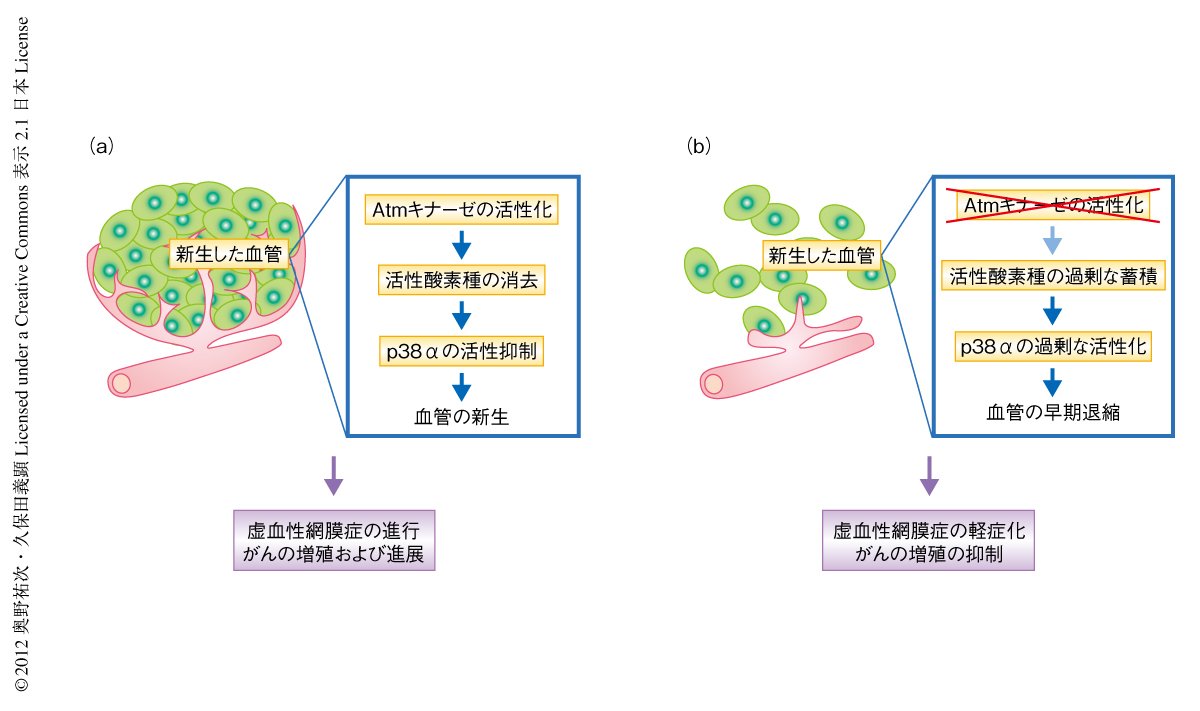
この病的な血管内皮において発現するAtmキナーゼの機能を調べるため,Atmノックアウトマウス,および,血管内皮に特異的なAtmノックアウトマウス7,8) に対し,虚血性網膜症モデルを施しその表現型を解析した.その結果,いずれのノックアウトマウスにおいても,野生型マウスにおいて旺盛に観察される病的な血管新生がほぼ消失していた(図1).また,Atmノックアウトマウスでは病的な血管新生における血管内皮細胞の増殖が減少する一方,アポトーシスが亢進しており,これが病的な血管新生の消失する原因と考えられた.マクロファージおよび好中球に特異的なAtmノックアウトマウス,あるいは,神経網膜に特異的なAtmノックアウトマウスも作製し,同様に虚血性網膜症モデルを施してその表現型を解析したが,野生型マウスと比べ有意な差は認められなかった.これらの結果から,Atmノックアウトマウスの虚血性網膜症モデルにおける表現型は,まぎれもなく血管内皮細胞におけるAtmキナーゼの機能喪失に起因するものと考えられた.

Atmノックアウトマウスにおける病的な血管新生の消失の分子機構を知るため,DNA損傷応答における下流シグナルとしてよく知られるヒストンH2AXおよびp53の活性化について検討した.Atmノックアウトマウスの血管内皮細胞において,ヒストンH2AXおよびp53のリン酸化のレベルは野生型マウスと比べ有意な差はなかった.また,p53ノックアウトマウスに虚血性網膜症モデルを施したが,Atmノックアウトマウスのような病的な血管新生の消失は観察されなかった.これらの結果は,Atmキナーゼの下流にあるDNA損傷応答とは別のシグナル伝達経路が,Atmノックアウトマウスにおける虚血性網膜症モデルの表現型において機能的に関与していることを示唆した.Atmノックアウトマウスの虚血性網膜症モデルにおける血管内皮細胞での活性酸素種の蓄積を2’,7’-ジクロロフルオレシンジアセタートによる染色により評価したところ,野生型マウスと比べ活性酸素種の蓄積は有意に亢進していた.活性酸素種の蓄積による血管内皮細胞へのダメージがAtmノックアウトマウスにおける病的な血管新生の消失をひき起こしているのではないかとの予想のもと,虚血性網膜症モデルを施したAtmノックアウトマウスに対し抗酸化剤N-アセチル-L-システインを処理した.その結果,病的な血管新生が野生型マウスと同じ程度に生じたことから,Atmキナーゼは活性酸素種の制御を介し病的な血管新生を促進していることが示された.
Atmキナーゼによる酸化ストレスの制御機構のそのさらに下流において,活性の変動するタンパク質を網羅的に探索した.その結果,p38 MAPキナーゼがAtmノックアウトマウスの血管内皮細胞において活性酸素種に依存してリン酸化の亢進していることを見い出した.虚血性網膜症モデルを施したAtmノックアウトマウスに対し,さきの抗酸化剤と同様に,p38 MAPキナーゼの阻害剤を処理したところ,病的な血管新生が野生型マウスと同じ程度に生じたことから,Atmキナーゼは酸化ストレスの制御機構の下流においてp38シグナル伝達経路の抑制を介して病的な血管新生を促進していることが示された.さらには,p38 MAPキナーゼのうちアポトーシス促進性のアイソフォームであるp38αにつき,血管内皮に特異的にAtm p38αダブルノックアウトマウス9) を作製したところ,虚血性網膜症モデルにおける病的な血管新生が野生型マウスと同じ程度に生じたことから,p38 MAPキナーゼのなかでもp38αがAtmキナーゼによる酸化ストレスの制御機構の下流において機能していることが示された.
Atmキナーゼの網膜での病的な血管新生における機能が,腫瘍での血管新生においても同様に機能しているかどうかを検討するため,B16メラノーマ細胞の皮下移植モデルを用いた検討を行った.正常な皮膚の血管内皮細胞とB16メラノーマに侵入する血管内皮細胞における活性酸素種の蓄積を2’,7’-ジクロロフルオレシンジアセタートによる染色により比較したところ,B16メラノーマに侵入する血管内皮細胞において有意に高かった.また,免疫染色の結果,B16メラノーマに侵入する血管内皮細胞ではリン酸化Atmキナーゼが検出されたが,正常な皮膚の血管内皮細胞では検出されなかった.これらの結果は,網膜と同様に,腫瘍においてもAtmキナーゼが病的な血管新生に対し選択的に機能していることを示唆していた.血管内皮に特異的なAtmノックアウトマウスにB16メラノーマ細胞を皮下移植したところ,腫瘍における血管新生および腫瘍の増大は著明に抑制された.血管内皮に特異的なAtm p38αダブルノックアウトマウスにおいては腫瘍における血管新生および腫瘍の増大は野生型マウスと同じ程度に生じたことから,Atmキナーゼによる酸化ストレスの制御機構の下流のp38αシグナル伝達経路は,腫瘍における血管新生においても機能していることが示された.さらには,血管内皮に特異的なAtmノックアウトマウスにB16メラノーマ細胞を皮下移植し,くわえてVEGFシグナルの阻害剤であるSU1498を投与したところ,Atm遺伝子の欠損とSU1498とが相乗的に腫瘍の抑制効果を示した.一方,生後30日目の成体のAtmノックアウトマウスの網膜,皮膚,気管,脂肪など健常な組織の血管構造にはまったく異常はみられなかったことから,Atmキナーゼによる血管新生作用は,VEGFシグナルとは異なり10),病的な血管新生に対し特異的であり健常な血管の維持には必要のないことが示唆された.
筆者らは,DNA損傷応答を介し腫瘍の形成に対する障壁として機能するAtmキナーゼが,一方では,酸化ストレスの制御機構を介し,腫瘍を含めた病的な血管新生に対し促進的に作用することをつきとめた.血管内皮に特異的なAtmノックアウトマウスではこの酸化ストレスの制御機構がはたらかず,活性酸素種が蓄積しp38αが過剰に活性されることで,病的な血管内皮は早期に退縮する(図2).この結果は,Atmキナーゼにおいても酸化ストレスの制御機構に寄与する部位を阻害する薬剤を開発することで,腫瘍における血管新生を選択的に傷害できる可能性を示唆する.また,がんの予防的な観点から有害とされてきた酸化ストレスが,腫瘍に栄養をあたえる血管新生を破綻させるという一面において,がんの病態および治療に有益であることを示唆する.
慶應義塾大学医学部 訪問研究員.
久保田 義顕(Yoshiaki Kubota)
略歴:2005年 慶應義塾大学大学院医学研究科 修了,同年 慶應義塾大学医学部 特別研究助手を経て,2009年より同 特任講師.
研究テーマ:発生期および病態における血管新生の制御機構.
抱負:兎角“大衆うけする研究”の求められるこのご時世,コテコテの発生・生化学研究に対する理解を,治療への応用を具体的に示すことでなんとか深めていきたい.
研究室URL:http://www.careerpath-prj.keio.ac.jp/kanrinmaru/scholar/kubota/
© 2012 奥野祐次・久保田義顕 Licensed under CC 表示 2.1 日本
(慶應義塾大学医学部 総合医科研究センター咸臨丸プロジェクト)
email:久保田義顕
DOI: 10.7875/first.author.2012.090
Pathological neoangiogenesis depends on oxidative stress regulation by ATM.
Yuji Okuno, Ayako Nakamura-Ishizu, Kinya Otsu, Toshio Suda, Yoshiaki Kubota
Nature Medicine, 18, 1208-1212 (2012)
要 約
AtmキナーゼはDNA損傷応答におけるマスター制御タンパク質であり,細胞の老化や腫瘍の形成に対する障壁として機能することが広く知られている.筆者らは,Atmキナーゼが活性酸素種の蓄積に応答し未成熟な血管において特異的に活性化されることを見い出した.マウスにおいて全身あるいは血管内皮に特異的にAtm遺伝子を欠損させると網膜における病的な血管新生が阻害された.この阻害はDNA損傷応答の異常ではなく,活性酸素種の増加およびp38αの過剰な活性化によりひき起こされていた.また,Atm遺伝子の欠損は腫瘍における血管新生の著明な減少,および,VEGFシグナルの阻害による抗血管新生作用の亢進もひき起こした.これらの結果は,病的な血管新生にはAtmキナーゼを介する酸化ストレスの制御機構が必要であること,また,活性酸素種の過剰な産生を促進する薬剤が血管新生病の治療に効果をもつ可能性を示唆した.
はじめに
生体のあらゆる組織がその恒常性を維持するためには,血管網から過不足なく酸素および栄養を供給されることが必須である.また,この組織への酸素供給に最適な血管のパターニングを規定する分子機構は,発生期における血管新生だけではなく,血管の過剰な増殖に端を発する血管新生病の病態の進行においても重要である.とくに,糖尿病性網膜症,加齢性黄斑変性症など,成人において失明の大きな原因のひとつとなる網膜血管新生病において,その病態の鍵となっている.さらには,がんの増大および進行においても,既存の血管からの血管新生がその病態に深くかかわっている.現在,臨床の場で用いられている抗がん剤の多くは腫瘍細胞の旺盛に増殖するという特徴を標的としたものであり,DNAやタンパク質の合成などの細胞分裂の過程を傷害するため,制がん効果の高いものほど正常組織,とくに,細胞のターンオーバーの速い造血細胞,消化管,皮膚などへのダメージも大きく,造血や消化管の障害など副作用が大きい.一方,腫瘍血管を標的とした“抗腫瘍血管新生療法”は,理論上,従来の抗がん剤に比べ正常な組織へのダメージは少なく,がんの内科的な治療におけるひとつのオプションとして確立されつつある.この抗腫瘍血管新生療法として中心的な戦略が,VEGF(vascular endothelial growth factor,血管内皮増殖因子)シグナルの阻害である.VEGFの中和抗体であるBevacizumabは,2004年の米国食品医薬品局,2007年の厚生労働省の認可ののち,臨床の場で広く用いられ,大腸がん,肺がん,腎がんなど多くのがん,とくに,ほかの内科的な治療に抵抗性の症例において有意な抗腫瘍効果をもつことが判明している1).ところが,一部のがんはこのVEGF阻害剤に対し耐性あるいは抵抗性を示し,とくに,VEGF阻害剤の投与を中断したのちに急激な血管の再成長をともなう腫瘍の増大をひき起こすことが判明しつつある2).さらには,VEGF阻害剤は腫瘍血管のみならず健常な血管をも傷害し,脳血管の障害や消化管の出血など,しばしば致命的な副作用を及ぼすことが報告されている3).これらを背景に,VEGFの阻害に代わる,または,併用しうる新たな抗血管新生療法の分子標的が世界的に探索されている.なかでも,腫瘍や網膜血管新生病における病的な血管のみに機能するタンパク質,つまり,病的な血管新生に選択的な治療標的が広く探索されている.
Atm(Ataxia telangiectasia mutated)遺伝子はヒトの毛細血管拡張性肉芽腫の原因遺伝子として知られている.Atm遺伝子のコードするAtmキナーゼはDNA損傷応答における中心的なタンパク質としてその機能はよく知られており,毛細血管拡張性肉芽腫の患者には高率(約30%)にがんが発生する4).しかしながら,毛細血管拡張性肉芽腫の患者に発生するがんは,ほとんどがリンパ腫や白血病などの腫瘍微小環境(がんニッチ)への依存性の低い造血系の腫瘍であり,固形腫瘍の発生はまれである5).これらの知見から,この研究は,Atmキナーゼの機能喪失による血管新生の異常がみかけ上,固形腫瘍の発生を覆い隠しているのではないかという仮説のもと,血管におけるAtmキナーゼの活性化および機能を解析した.
1.Atmキナーゼは酸化ストレスに応じ病的な血管において特異的に活性化される
平常時および病態における血管内皮細胞でのAtmキナーゼの活性化を検出するため,正常に発生したマウスの網膜,および,病的な血管新生のモデルである虚血性網膜症モデル6) を施したマウスの網膜に対し,Atmキナーゼ活性化の指標となる抗リン酸化Atmキナーゼ抗体による免疫染色を行った.その結果,リン酸化Atmキナーゼは正常な血管においてはほとんど検出されないものの,病的な血管新生において強く検出された.また,この病的な血管新生の生じた部位では血管内皮がジヒドロエチジウムにより強く染色され,活性酸素種の蓄積が示唆された.虚血性網膜症モデルを施したマウスを抗酸化剤であるN-アセチル-L-システインにより処理したところ,病的な血管新生の生じた部位におけるリン酸化Atmキナーゼの免疫染色の強度は,正常に発生した網膜の血管のレベルまで下がった.以上の結果から,Atmキナーゼはマウスの網膜において酸化ストレスに応じ病的な血管に特異的に活性化されていることが示された.
2.Atmキナーゼは網膜における病的な血管新生に必要である
この病的な血管内皮において発現するAtmキナーゼの機能を調べるため,Atmノックアウトマウス,および,血管内皮に特異的なAtmノックアウトマウス7,8) に対し,虚血性網膜症モデルを施しその表現型を解析した.その結果,いずれのノックアウトマウスにおいても,野生型マウスにおいて旺盛に観察される病的な血管新生がほぼ消失していた(図1).また,Atmノックアウトマウスでは病的な血管新生における血管内皮細胞の増殖が減少する一方,アポトーシスが亢進しており,これが病的な血管新生の消失する原因と考えられた.マクロファージおよび好中球に特異的なAtmノックアウトマウス,あるいは,神経網膜に特異的なAtmノックアウトマウスも作製し,同様に虚血性網膜症モデルを施してその表現型を解析したが,野生型マウスと比べ有意な差は認められなかった.これらの結果から,Atmノックアウトマウスの虚血性網膜症モデルにおける表現型は,まぎれもなく血管内皮細胞におけるAtmキナーゼの機能喪失に起因するものと考えられた.
3.Atmキナーゼは酸化ストレスの制御を介し病的な血管新生を促進する
Atmノックアウトマウスにおける病的な血管新生の消失の分子機構を知るため,DNA損傷応答における下流シグナルとしてよく知られるヒストンH2AXおよびp53の活性化について検討した.Atmノックアウトマウスの血管内皮細胞において,ヒストンH2AXおよびp53のリン酸化のレベルは野生型マウスと比べ有意な差はなかった.また,p53ノックアウトマウスに虚血性網膜症モデルを施したが,Atmノックアウトマウスのような病的な血管新生の消失は観察されなかった.これらの結果は,Atmキナーゼの下流にあるDNA損傷応答とは別のシグナル伝達経路が,Atmノックアウトマウスにおける虚血性網膜症モデルの表現型において機能的に関与していることを示唆した.Atmノックアウトマウスの虚血性網膜症モデルにおける血管内皮細胞での活性酸素種の蓄積を2’,7’-ジクロロフルオレシンジアセタートによる染色により評価したところ,野生型マウスと比べ活性酸素種の蓄積は有意に亢進していた.活性酸素種の蓄積による血管内皮細胞へのダメージがAtmノックアウトマウスにおける病的な血管新生の消失をひき起こしているのではないかとの予想のもと,虚血性網膜症モデルを施したAtmノックアウトマウスに対し抗酸化剤N-アセチル-L-システインを処理した.その結果,病的な血管新生が野生型マウスと同じ程度に生じたことから,Atmキナーゼは活性酸素種の制御を介し病的な血管新生を促進していることが示された.
4.Atmキナーゼは酸化ストレスの制御機構の下流においてp38シグナル伝達経路を抑制することで病的な血管新生を促進する
Atmキナーゼによる酸化ストレスの制御機構のそのさらに下流において,活性の変動するタンパク質を網羅的に探索した.その結果,p38 MAPキナーゼがAtmノックアウトマウスの血管内皮細胞において活性酸素種に依存してリン酸化の亢進していることを見い出した.虚血性網膜症モデルを施したAtmノックアウトマウスに対し,さきの抗酸化剤と同様に,p38 MAPキナーゼの阻害剤を処理したところ,病的な血管新生が野生型マウスと同じ程度に生じたことから,Atmキナーゼは酸化ストレスの制御機構の下流においてp38シグナル伝達経路の抑制を介して病的な血管新生を促進していることが示された.さらには,p38 MAPキナーゼのうちアポトーシス促進性のアイソフォームであるp38αにつき,血管内皮に特異的にAtm p38αダブルノックアウトマウス9) を作製したところ,虚血性網膜症モデルにおける病的な血管新生が野生型マウスと同じ程度に生じたことから,p38 MAPキナーゼのなかでもp38αがAtmキナーゼによる酸化ストレスの制御機構の下流において機能していることが示された.
5.Atmキナーゼは腫瘍における血管新生に必要である
Atmキナーゼの網膜での病的な血管新生における機能が,腫瘍での血管新生においても同様に機能しているかどうかを検討するため,B16メラノーマ細胞の皮下移植モデルを用いた検討を行った.正常な皮膚の血管内皮細胞とB16メラノーマに侵入する血管内皮細胞における活性酸素種の蓄積を2’,7’-ジクロロフルオレシンジアセタートによる染色により比較したところ,B16メラノーマに侵入する血管内皮細胞において有意に高かった.また,免疫染色の結果,B16メラノーマに侵入する血管内皮細胞ではリン酸化Atmキナーゼが検出されたが,正常な皮膚の血管内皮細胞では検出されなかった.これらの結果は,網膜と同様に,腫瘍においてもAtmキナーゼが病的な血管新生に対し選択的に機能していることを示唆していた.血管内皮に特異的なAtmノックアウトマウスにB16メラノーマ細胞を皮下移植したところ,腫瘍における血管新生および腫瘍の増大は著明に抑制された.血管内皮に特異的なAtm p38αダブルノックアウトマウスにおいては腫瘍における血管新生および腫瘍の増大は野生型マウスと同じ程度に生じたことから,Atmキナーゼによる酸化ストレスの制御機構の下流のp38αシグナル伝達経路は,腫瘍における血管新生においても機能していることが示された.さらには,血管内皮に特異的なAtmノックアウトマウスにB16メラノーマ細胞を皮下移植し,くわえてVEGFシグナルの阻害剤であるSU1498を投与したところ,Atm遺伝子の欠損とSU1498とが相乗的に腫瘍の抑制効果を示した.一方,生後30日目の成体のAtmノックアウトマウスの網膜,皮膚,気管,脂肪など健常な組織の血管構造にはまったく異常はみられなかったことから,Atmキナーゼによる血管新生作用は,VEGFシグナルとは異なり10),病的な血管新生に対し特異的であり健常な血管の維持には必要のないことが示唆された.
おわりに
筆者らは,DNA損傷応答を介し腫瘍の形成に対する障壁として機能するAtmキナーゼが,一方では,酸化ストレスの制御機構を介し,腫瘍を含めた病的な血管新生に対し促進的に作用することをつきとめた.血管内皮に特異的なAtmノックアウトマウスではこの酸化ストレスの制御機構がはたらかず,活性酸素種が蓄積しp38αが過剰に活性されることで,病的な血管内皮は早期に退縮する(図2).この結果は,Atmキナーゼにおいても酸化ストレスの制御機構に寄与する部位を阻害する薬剤を開発することで,腫瘍における血管新生を選択的に傷害できる可能性を示唆する.また,がんの予防的な観点から有害とされてきた酸化ストレスが,腫瘍に栄養をあたえる血管新生を破綻させるという一面において,がんの病態および治療に有益であることを示唆する.
文 献
- Carmeliet, P. & Jain, R. K.: Molecular mechanisms and clinical applications of angiogenesis. Nature, 473, 298-307 (2011)[PubMed]
- Mancuso, M. R., Davis, R., Norberg, S. M. et al.: Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J. Clin. Invest., 116, 2610-2621 (2006)[PubMed]
- Bergers, G. & Hanahan, D.: Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer, 8, 592-603 (2008)[PubMed]
- Shiloh, Y.: ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer, 3, 155-168 (2003)[PubMed]
- McKinnon, P. J.: ATM and the molecular pathogenesis of ataxia telangiectasia. Annu. Rev. Pathol., 7, 303-321 (2012)[PubMed]
- Smith, L. E., Wesolowski, E., McLellan, A. et al.: Oxygen-induced retinopathy in the mouse. Invest. Ophthalmol. Vis. Sci., 35, 101-111 (1994)[PubMed]
- Herzog, K. H., Chong, M. J., Kapsetaki, M. et al.: Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science, 280, 1089-1091 (1998)[PubMed]
- Zha, S., Jiang, W., Fujiwara, Y. et al.: Ataxia telangiectasia-mutated protein and DNA-dependent protein kinase have complementary V(D)J recombination functions. Proc. Natl. Acad. Sci. USA, 108, 2028-2033 (2011)[PubMed]
- Nishida, K., Yamaguchi, O., Hirotani, S. et al.: p38α mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol. Cell. Biol., 24, 10611-10620 (2004)[PubMed]
- Kubota, Y., Takubo, K., Shimizu, T. et al.: M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J. Exp. Med., 206, 1089-1102 (2009)[PubMed]
著者プロフィール
慶應義塾大学医学部 訪問研究員.
久保田 義顕(Yoshiaki Kubota)
略歴:2005年 慶應義塾大学大学院医学研究科 修了,同年 慶應義塾大学医学部 特別研究助手を経て,2009年より同 特任講師.
研究テーマ:発生期および病態における血管新生の制御機構.
抱負:兎角“大衆うけする研究”の求められるこのご時世,コテコテの発生・生化学研究に対する理解を,治療への応用を具体的に示すことでなんとか深めていきたい.
研究室URL:http://www.careerpath-prj.keio.ac.jp/kanrinmaru/scholar/kubota/
© 2012 奥野祐次・久保田義顕 Licensed under CC 表示 2.1 日本
