転写共役因子CITED2はホルモンに応答し転写コアクチベーターPGC-1αのアセチル化を調節して肝臓における糖新生を制御する
酒井真志人・松本道宏・春日雅人
(国立国際医療研究センター研究所 糖尿病研究センター分子代謝制御研究部)
email:酒井真志人,松本道宏
DOI: 10.7875/first.author.2012.045
CITED2 links hormonal signaling to PGC-1α acetylation in regulation of gluconeogenesis.
Mashito Sakai, Michihiro Matsumoto, Tomoko Tujimura, Cao Yongheng, Tetsuya Noguchi, Kenjiro Inagaki, Hiroshi Inoue, Tetsuya Hosooka, Kazuo Takazawa, Yoshiaki Kido, Kazuki Yasuda, Ryuji Hiramatsu, Yasushi Matsuki, Masato Kasuga
Nature Medicine, 18, 612-617 (2012)
2型糖尿病では肝臓における糖新生の亢進により糖の産生が増加しており,高血糖の一因となっている.したがって,糖新生の制御機構の解明は高血糖の治療薬の開発にあたり重要な課題である.今回,筆者らは,グルカゴンにより発現の誘導される転写共役因子CITED2が転写コアクチベーターPGC-1αを活性化し,絶食時の糖新生系の酵素の発現を上昇させることを見い出した.CITED2によるPGC-1αの活性化は,PGC-1αをアセチル化により不活性化するGCN5とCITED2とが複合体を形成し,PGC-1αのアセチル化を抑制することにより起こることも明らかにした.また,インスリンがCITED2とGCN5との相互作用を阻害し,GCN5によるPGC-1αのアセチル化を促進して糖新生を抑制することも明らかにした.肥満および糖尿病モデルマウスであるdb/dbマウスの肝臓ではCITED2の発現が著明に上昇しており,CITED2のノックダウンにより糖新生系の酵素の発現が低下し高血糖が改善した.この研究により,CITED2はグルカゴンおよびインスリンの刺激に応答してPGC-1αのアセチル化および活性を介し糖新生を制御する重要な代謝制御タンパク質であることが明らかになり,CITED2の作用の抑制が高血糖に対する治療標的となりうることが示された.
2型糖尿病における高血糖の主要な原因のひとつとして,インスリン抵抗性にともなう肝臓からの糖の産生の病的な亢進があげられ1),その分子機構の解明は新規の糖尿病治療薬の開発のため重要な課題である.転写共役因子CITED2はヒストンアセチル化酵素活性をもつ転写コアクチベーターCBP/p300との相互作用を介し転写を制御することが報告されていた2).また,ノックアウトマウスを用いた検討から,CITED2は胎仔の発生や3),成体における造血幹細胞の維持4) に不可欠であることが明らかになっていた.一方,CITED2はHNF-4α5),PPARα/γ6),CBPといった肝臓における糖脂質の代謝制御において重要な役割をはたすタンパク質と相互作用することが報告されていたが,肝臓での糖代謝の制御における役割は明らかになっていなかった.筆者らは,肝臓におけるCITED2の量が摂食状態の変化により制御されていることを見い出し,肝臓での糖代謝の制御におけるCITED2の役割を検討した.
マウスの肝臓におけるCITED2の量は,絶食時ならびにグルカゴンの腹腔内への投与時に増加した.また,培養肝細胞における検討では,CITED2はグルカゴンあるいはそのセカンドメッセンジャーであるcAMPによる処理により増加し,その増加はプロテインキナーゼA阻害剤による処理により抑制された.このことから,CITED2の発現はグルカゴン-cAMP-プロテインキナーゼAシグナル伝達経路により上昇することが明らかになった.CITED2はユビキチン-プロテアソーム系により急速に分解されることが報告されている2,7).肝細胞においてCITED2を免疫沈降しそのユビキチン化を検討したところcAMP処理によりユビキチン化の低下がみられ,CITED2の増加の少なくとも一部はユビキチン化の低下による分解抑制によるものと考えられた.
CITED2が絶食時の肝臓における糖代謝の制御に関与する可能性を考え,初代培養肝細胞において,cAMPの刺激により糖新生系の酵素の発現誘導および糖の産生に対するCITED2のノックダウンならびに過剰発現の効果を検討した.CITED2のshRNAを発現するアデノウイルスによりCITED2をノックダウンすると,cAMP刺激による糖新生系の酵素をコードするG6pc遺伝子およびPck1遺伝子の発現誘導は障害され糖の産生は減少した.逆に,CITED2の過剰発現によりG6pc遺伝子およびPck1遺伝子の発現は上昇し糖の産生は著明に増強した.この増強作用はCBPならびにHNF-4αへの結合に必要な部位であるCR2ドメインを欠損したCITED2変異体の過剰発現によっても認められたことから,CITED2によるcAMPに依存性の糖新生系酵素の発現の制御にはCBPあるいはHNF4αとの結合は必要としないことが示唆された.これらの結果から,CITED2はin vitroでは糖新生系酵素の発現制御を介して糖の産生を制御していると考えられたので,つぎに,in vivoにおける効果を検討した.マウスの肝臓においてアデノウイルスベクターを用いてCITED2を過剰発現すると,絶食時においてG6pc遺伝子およびPck1遺伝子の発現が約3倍に上昇し,空腹時の血糖値が上昇した.また,肝臓からの糖の新生能を評価するピルビン酸負荷試験を行ったところ,CITED2の過剰発現によりピルビン酸の負荷後の血糖の上昇が著明に増加し,CITED2がin vivoでも肝臓における糖新生を増加させることが明らかになった.
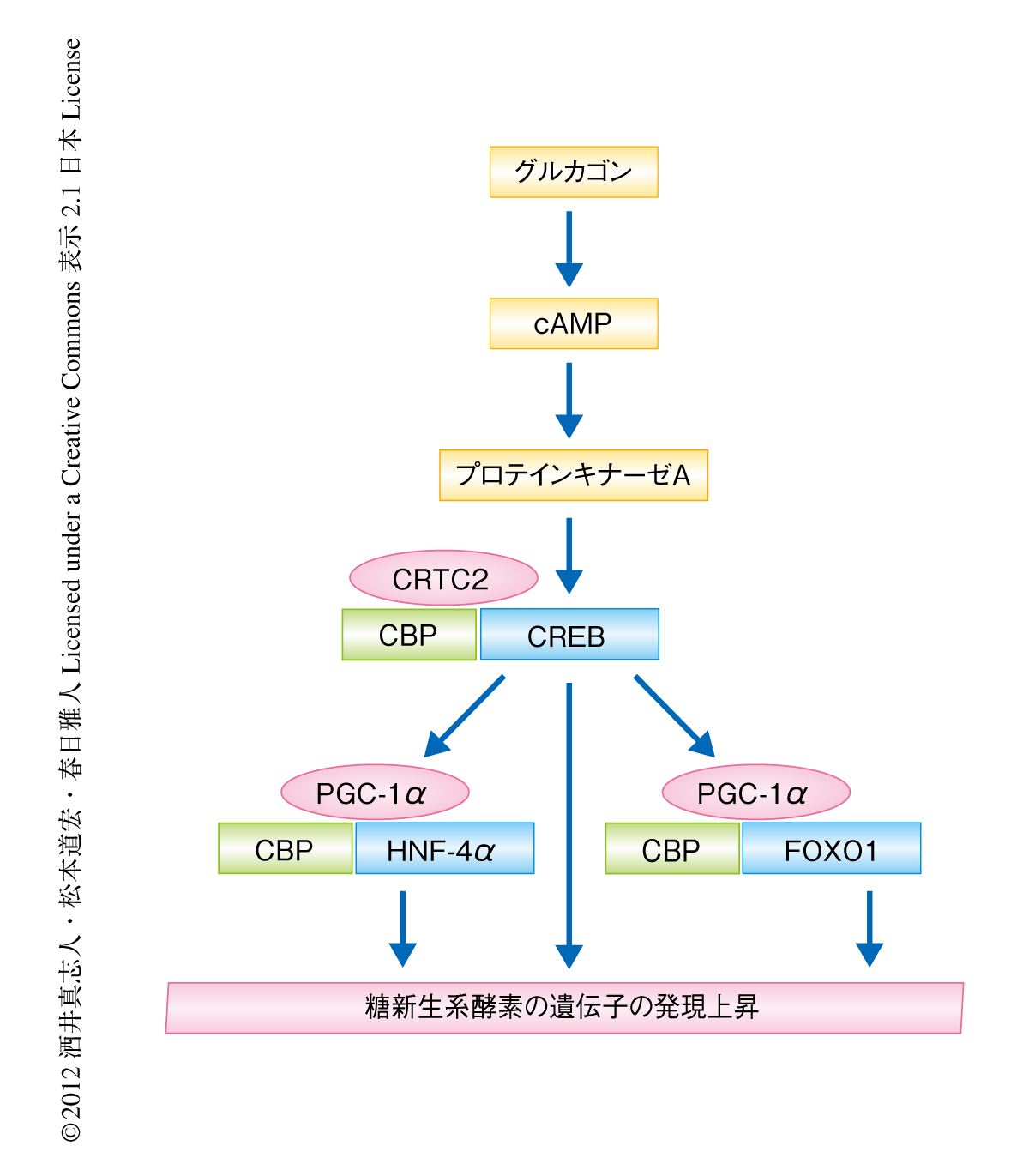
CITED2が肝臓において糖新生系酵素の発現を介し血糖値を制御していると考えられたので,その分子機構について検討した.転写コアクチベーターであるPGC-1αは絶食時にグルカゴン-cAMP-プロテインキナーゼAシグナル伝達経路によりその発現が上昇し糖新生系酵素の発現を強力に誘導する,肝臓における糖の産生の鍵タンパク質である8)(図1).そこで,CITED2によるcAMPに依存性の糖新生系酵素の発現誘導におけるCITED2の効果がPGC-1αを介しているかどうか検討したところ,PGC-1αをノックダウンした初代培養肝細胞ではCITED2の効果は消失していた.PGC-1αに依存性の糖新生系酵素の発現誘導へのCITED2の関与についても検討したところ,PGC-1αによる発現誘導はCITED2のノックダウンにより著明に抑制され,逆に,CITED2の過剰発現により増強した.さらに,CITED2ならびにCR2ドメインを欠損したCITED2変異体についてPGC-1αのコアクチベーター活性への影響をG6pc遺伝子プロモーターアッセイにより検討したところ,いずれもコアクチベーター活性を増強した.これらの結果より,CITED2はCR2ドメインに非依存性にPGC-1αのコアクチベーター活性を増強することが示唆された.
CITED2によるPGC-1αの活性増強の分子機構を検討した.PGC-1αの活性はアセチル化,リン酸化,メチル化,SUMO化などさまざまな翻訳後修飾により制御されている.PGC-1αのアセチル化に対するCITED2の効果を検討したところ,CITED2はPGC-1αのアセチル化を抑制することが明らかになった.PGC-1αの活性はアセチル化酵素GCN5によるアセチル化により抑制され9),脱アセチル化酵素SIRT1による脱アセチル化により増強する10) ことが報告されている.そこで,CITED2とPGC-1α,SIRT1,GCN5との相互作用について共沈実験により検討したところ,CITED2はPGC-1αおよびSIRT1のいずれとも共沈しなかった.CITED2はPGC-1αとSIRT1との結合や,細胞内におけるNAD+とNADHとの比,および,SIRT1のターゲットであるFOXO1のアセチル化には影響をあたえず,SIRT1に依存性のPGC-1αの脱アセチル化には関与しないものと考えられた.一方,CITED2はGCN5と共沈しこれと相互作用することが示唆された.そこで,HEK293細胞においてGCN5によるPGC-1αのアセチル化に対するCITED2の効果を検討した.すでに報告されていたように,GCN5はPGC-1αに結合しそのアセチル化を増加させたが,CITED2はPGC-1αに結合するGCN5ならびにPGC-1αのアセチル化を用量に依存性に減少させた.一方,GCN5のヒストンアセチル化活性に対するCITED2の効果をヒストンH3を基質としたHATアッセイにより検討したところ,CITED2の過剰発現はGCN5の活性を抑制しないことが明らかになった.これらの結果から,CITED2はGCN5と結合し,PGC-1αとGCN5との結合を阻害することでPGC-1αのアセチル化を抑制するものと考えられた.
CITED2とGCN5との相互作用に必要な部位を同定するため,それぞれの欠損変異体をHEK293細胞に発現させ共沈実験を行った.その結果,CITED2のCR1ドメインおよびSRJドメインがGCN5との相互作用に必要であることが明らかになった.実際,SRJドメインを欠損したCITED2変異体はGCN5に依存性のPGC-1αのアセチル化を抑制せず,初代培養肝細胞においてcAMPに依存性の糖新生系酵素の発現を上昇させなかった.
CITED2とGCN5との相互作用にインスリンのあたえる影響を検討した.インスリンはGCN5と共沈するCITED2の量を減少させ,この効果はPI3キナーゼ阻害剤およびAkt阻害剤で解除されたが,mTORの阻害剤であるラパマイシンでは解除されなかった.また,インスリンはCITED2によるPGC-1αのアセチル化の抑制を完全に回復させた.これらの結果から,インスリンはPI3キナーゼ-Aktシグナル伝達経路を介してCITED2とGCN5との相互作用を低下させ,PGC-1αのアセチル化を回復させることが明らかになった.
db/dbマウスはレプチン受容体の変異のため多食,肥満,糖尿病を呈する.db/dbマウスや高脂肪食負荷マウスは肥満および糖尿病のモデルマウスとして有用である.これらのモデルマウスの肝臓におけるCITED2 mRNAおよびCITED2タンパク質の発現を検討したところ,いずれも著明に上昇していた.さらに,db/dbマウスの肝臓においてCITED2をノックダウンすると,糖新生系酵素をコードするG6pc遺伝子およびPck1遺伝子の発現が大きく低下し高血糖が改善した.これらの結果から,CITED2がdb/dbマウスの病的な糖の産生の亢進に強く関与すること,また,糖尿病モデルマウスの肝臓におけるCITED2の作用の抑制により高血糖の治療ができること,が示唆された.
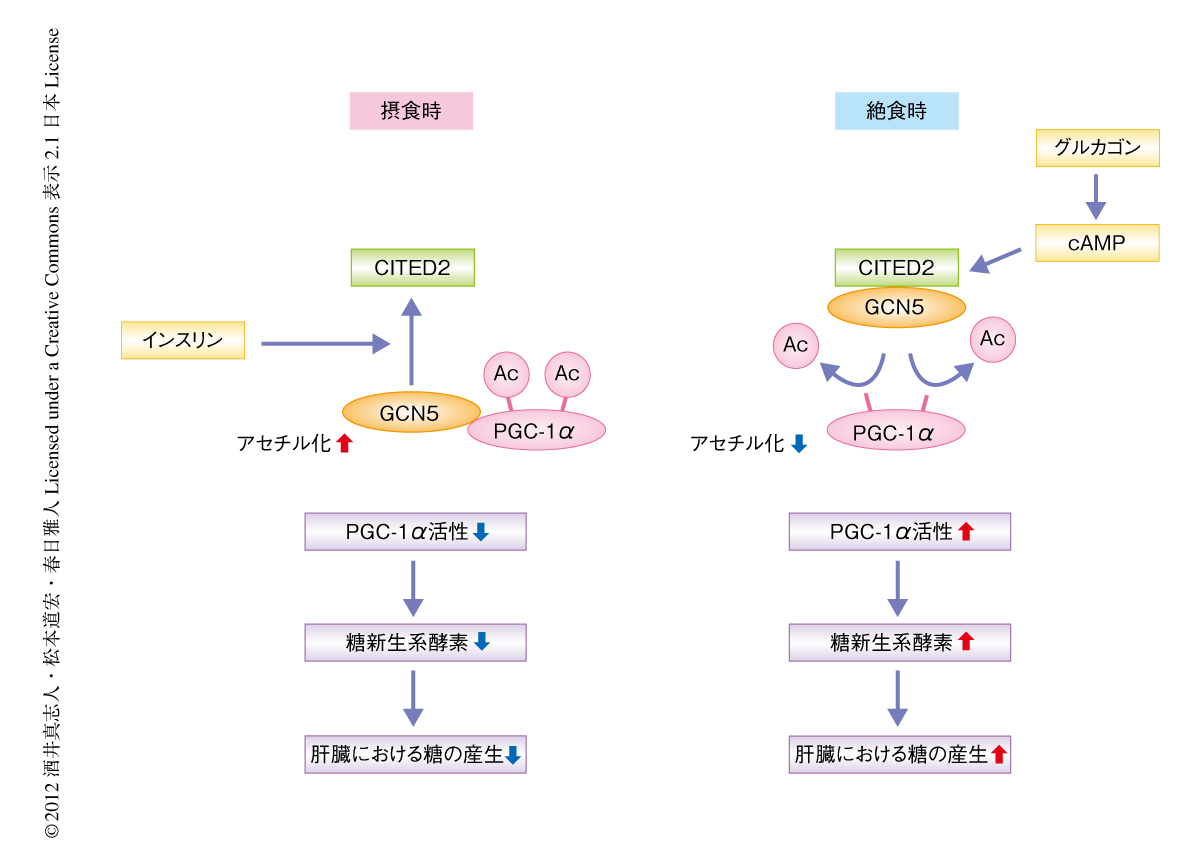
この研究により,転写共役因子CITED2による肝臓における新たな糖の産生の制御機構が明らかになった.摂食時にはグルカゴンのレベルの低下によりCITED2の発現が低下するとともに,インスリンによりCITED2によるGCN5の抑制が解除され,GCN5によるPGC-1αのアセチル化は亢進し,PGC-1αは不活化され糖の産生は抑制される.一方,絶食時にはグルカゴンによりCITED2の発現が上昇し,GCN5によるPGC-1αのアセチル化を抑制することで,PGC-1αの活性は上昇し糖の産生は増加する(図2).CITED2はグルカゴンおよびインスリンの刺激に応答してPGC-1αのアセチル化および活性を介し糖新生を制御する重要な代謝制御タンパク質である.また,肥満および糖尿病のモデルマウスであるdb/dbマウス,あるいは,高脂肪食負荷マウスの肝臓ではCITED2の発現の上昇が糖の産生亢進の一因であり,CITED2の作用の抑制が高血糖の治療につながる可能性がある.新規の糖尿病治療薬の標的分子としてのCITED2抑制因子の同定は今後の重要な課題であると考える.
略歴:2012年 神戸大学大学院医学研究科 修了,2010年より国立国際医療研究センター研究所 研究員.
研究テーマ:糖脂質代謝における転写制御.
抱負:エネルギー代謝制御とその破綻の分子機構の解明に貢献したい.
松本 道宏(Michihiro Matsumoto)
国立国際医療研究センター研究所 部長.
春日 雅人(Masato Kasuga)
国立国際医療研究センター 理事長.
© 2012 酒井真志人・松本道宏・春日雅人 Licensed under CC 表示 2.1 日本
(国立国際医療研究センター研究所 糖尿病研究センター分子代謝制御研究部)
email:酒井真志人,松本道宏
DOI: 10.7875/first.author.2012.045
CITED2 links hormonal signaling to PGC-1α acetylation in regulation of gluconeogenesis.
Mashito Sakai, Michihiro Matsumoto, Tomoko Tujimura, Cao Yongheng, Tetsuya Noguchi, Kenjiro Inagaki, Hiroshi Inoue, Tetsuya Hosooka, Kazuo Takazawa, Yoshiaki Kido, Kazuki Yasuda, Ryuji Hiramatsu, Yasushi Matsuki, Masato Kasuga
Nature Medicine, 18, 612-617 (2012)
要 約
2型糖尿病では肝臓における糖新生の亢進により糖の産生が増加しており,高血糖の一因となっている.したがって,糖新生の制御機構の解明は高血糖の治療薬の開発にあたり重要な課題である.今回,筆者らは,グルカゴンにより発現の誘導される転写共役因子CITED2が転写コアクチベーターPGC-1αを活性化し,絶食時の糖新生系の酵素の発現を上昇させることを見い出した.CITED2によるPGC-1αの活性化は,PGC-1αをアセチル化により不活性化するGCN5とCITED2とが複合体を形成し,PGC-1αのアセチル化を抑制することにより起こることも明らかにした.また,インスリンがCITED2とGCN5との相互作用を阻害し,GCN5によるPGC-1αのアセチル化を促進して糖新生を抑制することも明らかにした.肥満および糖尿病モデルマウスであるdb/dbマウスの肝臓ではCITED2の発現が著明に上昇しており,CITED2のノックダウンにより糖新生系の酵素の発現が低下し高血糖が改善した.この研究により,CITED2はグルカゴンおよびインスリンの刺激に応答してPGC-1αのアセチル化および活性を介し糖新生を制御する重要な代謝制御タンパク質であることが明らかになり,CITED2の作用の抑制が高血糖に対する治療標的となりうることが示された.
はじめに
2型糖尿病における高血糖の主要な原因のひとつとして,インスリン抵抗性にともなう肝臓からの糖の産生の病的な亢進があげられ1),その分子機構の解明は新規の糖尿病治療薬の開発のため重要な課題である.転写共役因子CITED2はヒストンアセチル化酵素活性をもつ転写コアクチベーターCBP/p300との相互作用を介し転写を制御することが報告されていた2).また,ノックアウトマウスを用いた検討から,CITED2は胎仔の発生や3),成体における造血幹細胞の維持4) に不可欠であることが明らかになっていた.一方,CITED2はHNF-4α5),PPARα/γ6),CBPといった肝臓における糖脂質の代謝制御において重要な役割をはたすタンパク質と相互作用することが報告されていたが,肝臓での糖代謝の制御における役割は明らかになっていなかった.筆者らは,肝臓におけるCITED2の量が摂食状態の変化により制御されていることを見い出し,肝臓での糖代謝の制御におけるCITED2の役割を検討した.
1.肝臓におけるCITED2の発現はグルカゴンにより上昇する
マウスの肝臓におけるCITED2の量は,絶食時ならびにグルカゴンの腹腔内への投与時に増加した.また,培養肝細胞における検討では,CITED2はグルカゴンあるいはそのセカンドメッセンジャーであるcAMPによる処理により増加し,その増加はプロテインキナーゼA阻害剤による処理により抑制された.このことから,CITED2の発現はグルカゴン-cAMP-プロテインキナーゼAシグナル伝達経路により上昇することが明らかになった.CITED2はユビキチン-プロテアソーム系により急速に分解されることが報告されている2,7).肝細胞においてCITED2を免疫沈降しそのユビキチン化を検討したところcAMP処理によりユビキチン化の低下がみられ,CITED2の増加の少なくとも一部はユビキチン化の低下による分解抑制によるものと考えられた.
2.CITED2は肝臓において糖新生系の酵素の発現と糖の産生を上昇させる
CITED2が絶食時の肝臓における糖代謝の制御に関与する可能性を考え,初代培養肝細胞において,cAMPの刺激により糖新生系の酵素の発現誘導および糖の産生に対するCITED2のノックダウンならびに過剰発現の効果を検討した.CITED2のshRNAを発現するアデノウイルスによりCITED2をノックダウンすると,cAMP刺激による糖新生系の酵素をコードするG6pc遺伝子およびPck1遺伝子の発現誘導は障害され糖の産生は減少した.逆に,CITED2の過剰発現によりG6pc遺伝子およびPck1遺伝子の発現は上昇し糖の産生は著明に増強した.この増強作用はCBPならびにHNF-4αへの結合に必要な部位であるCR2ドメインを欠損したCITED2変異体の過剰発現によっても認められたことから,CITED2によるcAMPに依存性の糖新生系酵素の発現の制御にはCBPあるいはHNF4αとの結合は必要としないことが示唆された.これらの結果から,CITED2はin vitroでは糖新生系酵素の発現制御を介して糖の産生を制御していると考えられたので,つぎに,in vivoにおける効果を検討した.マウスの肝臓においてアデノウイルスベクターを用いてCITED2を過剰発現すると,絶食時においてG6pc遺伝子およびPck1遺伝子の発現が約3倍に上昇し,空腹時の血糖値が上昇した.また,肝臓からの糖の新生能を評価するピルビン酸負荷試験を行ったところ,CITED2の過剰発現によりピルビン酸の負荷後の血糖の上昇が著明に増加し,CITED2がin vivoでも肝臓における糖新生を増加させることが明らかになった.
3.CITED2はPGC-1αのコアクチベーター活性を上昇させる
CITED2が肝臓において糖新生系酵素の発現を介し血糖値を制御していると考えられたので,その分子機構について検討した.転写コアクチベーターであるPGC-1αは絶食時にグルカゴン-cAMP-プロテインキナーゼAシグナル伝達経路によりその発現が上昇し糖新生系酵素の発現を強力に誘導する,肝臓における糖の産生の鍵タンパク質である8)(図1).そこで,CITED2によるcAMPに依存性の糖新生系酵素の発現誘導におけるCITED2の効果がPGC-1αを介しているかどうか検討したところ,PGC-1αをノックダウンした初代培養肝細胞ではCITED2の効果は消失していた.PGC-1αに依存性の糖新生系酵素の発現誘導へのCITED2の関与についても検討したところ,PGC-1αによる発現誘導はCITED2のノックダウンにより著明に抑制され,逆に,CITED2の過剰発現により増強した.さらに,CITED2ならびにCR2ドメインを欠損したCITED2変異体についてPGC-1αのコアクチベーター活性への影響をG6pc遺伝子プロモーターアッセイにより検討したところ,いずれもコアクチベーター活性を増強した.これらの結果より,CITED2はCR2ドメインに非依存性にPGC-1αのコアクチベーター活性を増強することが示唆された.
4.CITED2はGCN5によるPGC-1αのアセチル化を抑制する
CITED2によるPGC-1αの活性増強の分子機構を検討した.PGC-1αの活性はアセチル化,リン酸化,メチル化,SUMO化などさまざまな翻訳後修飾により制御されている.PGC-1αのアセチル化に対するCITED2の効果を検討したところ,CITED2はPGC-1αのアセチル化を抑制することが明らかになった.PGC-1αの活性はアセチル化酵素GCN5によるアセチル化により抑制され9),脱アセチル化酵素SIRT1による脱アセチル化により増強する10) ことが報告されている.そこで,CITED2とPGC-1α,SIRT1,GCN5との相互作用について共沈実験により検討したところ,CITED2はPGC-1αおよびSIRT1のいずれとも共沈しなかった.CITED2はPGC-1αとSIRT1との結合や,細胞内におけるNAD+とNADHとの比,および,SIRT1のターゲットであるFOXO1のアセチル化には影響をあたえず,SIRT1に依存性のPGC-1αの脱アセチル化には関与しないものと考えられた.一方,CITED2はGCN5と共沈しこれと相互作用することが示唆された.そこで,HEK293細胞においてGCN5によるPGC-1αのアセチル化に対するCITED2の効果を検討した.すでに報告されていたように,GCN5はPGC-1αに結合しそのアセチル化を増加させたが,CITED2はPGC-1αに結合するGCN5ならびにPGC-1αのアセチル化を用量に依存性に減少させた.一方,GCN5のヒストンアセチル化活性に対するCITED2の効果をヒストンH3を基質としたHATアッセイにより検討したところ,CITED2の過剰発現はGCN5の活性を抑制しないことが明らかになった.これらの結果から,CITED2はGCN5と結合し,PGC-1αとGCN5との結合を阻害することでPGC-1αのアセチル化を抑制するものと考えられた.
CITED2とGCN5との相互作用に必要な部位を同定するため,それぞれの欠損変異体をHEK293細胞に発現させ共沈実験を行った.その結果,CITED2のCR1ドメインおよびSRJドメインがGCN5との相互作用に必要であることが明らかになった.実際,SRJドメインを欠損したCITED2変異体はGCN5に依存性のPGC-1αのアセチル化を抑制せず,初代培養肝細胞においてcAMPに依存性の糖新生系酵素の発現を上昇させなかった.
5.CITED2によるPGC-1αアセチル化の抑制はインスリンにより解除される
CITED2とGCN5との相互作用にインスリンのあたえる影響を検討した.インスリンはGCN5と共沈するCITED2の量を減少させ,この効果はPI3キナーゼ阻害剤およびAkt阻害剤で解除されたが,mTORの阻害剤であるラパマイシンでは解除されなかった.また,インスリンはCITED2によるPGC-1αのアセチル化の抑制を完全に回復させた.これらの結果から,インスリンはPI3キナーゼ-Aktシグナル伝達経路を介してCITED2とGCN5との相互作用を低下させ,PGC-1αのアセチル化を回復させることが明らかになった.
6.CITED2の発現抑制により糖尿病のモデルマウスの高血糖が改善する
db/dbマウスはレプチン受容体の変異のため多食,肥満,糖尿病を呈する.db/dbマウスや高脂肪食負荷マウスは肥満および糖尿病のモデルマウスとして有用である.これらのモデルマウスの肝臓におけるCITED2 mRNAおよびCITED2タンパク質の発現を検討したところ,いずれも著明に上昇していた.さらに,db/dbマウスの肝臓においてCITED2をノックダウンすると,糖新生系酵素をコードするG6pc遺伝子およびPck1遺伝子の発現が大きく低下し高血糖が改善した.これらの結果から,CITED2がdb/dbマウスの病的な糖の産生の亢進に強く関与すること,また,糖尿病モデルマウスの肝臓におけるCITED2の作用の抑制により高血糖の治療ができること,が示唆された.
おわりに
この研究により,転写共役因子CITED2による肝臓における新たな糖の産生の制御機構が明らかになった.摂食時にはグルカゴンのレベルの低下によりCITED2の発現が低下するとともに,インスリンによりCITED2によるGCN5の抑制が解除され,GCN5によるPGC-1αのアセチル化は亢進し,PGC-1αは不活化され糖の産生は抑制される.一方,絶食時にはグルカゴンによりCITED2の発現が上昇し,GCN5によるPGC-1αのアセチル化を抑制することで,PGC-1αの活性は上昇し糖の産生は増加する(図2).CITED2はグルカゴンおよびインスリンの刺激に応答してPGC-1αのアセチル化および活性を介し糖新生を制御する重要な代謝制御タンパク質である.また,肥満および糖尿病のモデルマウスであるdb/dbマウス,あるいは,高脂肪食負荷マウスの肝臓ではCITED2の発現の上昇が糖の産生亢進の一因であり,CITED2の作用の抑制が高血糖の治療につながる可能性がある.新規の糖尿病治療薬の標的分子としてのCITED2抑制因子の同定は今後の重要な課題であると考える.
文 献
- Biddinger, S. B. & Kahn, C. R.: From mice to men: insights into the insulin resistance syndromes. Annu. Rev. Physiol., 68, 123-158 (2006)[PubMed]
- Bhattacharya, S., Michels, C. L., Leung, M. K. et al.: Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1. Genes Dev., 13, 64-75 (1999)[PubMed]
- Bamforth, S. D., Braganca, J., Eloranta, J. J. et al.: Cardiac malformations, adrenal agenesis, neural crest defects and exencephaly in mice lacking Cited2, a new Tfap2 co-activator. Nat. Genet., 29, 469-474 (2001)[PubMed]
- Kranc, K. R., Schepers, H, Rodrigues, N. P. et al.: Cited2 is an essential regulator of adult hematopoietic stem cells. Cell Stem Cell, 5, 659-665 (2009)[PubMed]
- Qu, X., Lam, E., Doughman, Y. Q. et al.: Cited2, a coactivator of HNF4α, is essential for liver development. EMBO J., 26, 4445-4456 (2007)[PubMed]
- Tien, E. S., Davis, J. W. & Vanden Heuvel, J. P.: Identification of the CREB-binding protein/p300-interacting protein CITED2 as a peroxisome proliferator-activated receptor α coregulator. J. Biol. Chem., 279, 24053-24063 (2004)[PubMed]
- Sin, D. H., Li, S. H., Chun. Y. S. et al.: CITED2 mediates the paradoxical responses of HIF-1α to proteasome inhibition. Oncogene, 27, 1939-1944 (2008)[PubMed]
- Lin, J., Handschin, C. & Spiegelman, B. M.: Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab., 1, 361-370 (2005)[PubMed]
- Lerin, C., Rodgers, J. T., Kalume, D. E. et al.: GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1α. Cell Metab., 3, 429-438 (2006)[PubMed]
- Rodgers, J. T., Lerin, C., Haas, W. et al.: Nutrient control of glucose homeostasis through a complex of PGC-1αand SIRT1. Nature, 434, 113-118 (2005)[PubMed]
著者プロフィール
略歴:2012年 神戸大学大学院医学研究科 修了,2010年より国立国際医療研究センター研究所 研究員.
研究テーマ:糖脂質代謝における転写制御.
抱負:エネルギー代謝制御とその破綻の分子機構の解明に貢献したい.
松本 道宏(Michihiro Matsumoto)
国立国際医療研究センター研究所 部長.
春日 雅人(Masato Kasuga)
国立国際医療研究センター 理事長.
© 2012 酒井真志人・松本道宏・春日雅人 Licensed under CC 表示 2.1 日本
