チロシンキナーゼBtkはヒトの好中球において活性酸素種の産生および刺激により誘導されるアポトーシスを負に制御する
本田富美子・森尾友宏
(東京医科歯科大学大学院医歯学総合研究科 発生発達病態学)
email:本田富美子,森尾友宏
DOI: 10.7875/first.author.2012.036
The kinase Btk negatively regulates the production of reactive oxygen species and stimulation-induced apoptosis in human neutrophils.
Fumiko Honda, Hirotsugu Kano, Hirokazu Kanegane, Shigeaki Nonoyama, Eun-Sung Kim, Sang-Kyou Lee, Masatoshi Takagi, Shuki Mizutani, Tomohiro Morio
Nature Immunology, 13, 369-378 (2012)
X連鎖無γグロブリン血症はBTK遺伝子の異常によりB細胞の欠損する免疫不全症である.この疾患では軽い細菌感染症によっても好中球の減少が起こり,場合によっては生命の危険になることが知られている.この研究において,筆者らは,BTK遺伝子によりコードされているチロシンキナーゼBtkが好中球において特別のはたらきをしていて,刺激の入るまえにはMalが細胞膜へと移動するのを抑え,好中球による活性酸素種の産生におけるプライミングの段階を微調整し,軽い刺激に反応して活性酸素種を過剰に産生しないよう制御していることを明らかにした.また,好中球においてMalを制御する詳細なシグナル伝達機構をはじめて示した.さらに,好中球にBtkを効率よく導入したりその機能を抑えたりすることで,好中球からの活性酸素種の産生やアポトーシスを制御できることがわかった.
好中球は微生物の感染の初期に動員され貪食や殺傷を行う,生体防御において重要な細胞である1).取り込んだ病原体はさまざまな機構により殺傷されるが,その中心的な役割をするのが活性酸素種である.活性酸素種は細胞毒性をもち自らの細胞を傷つけるなどのおそれがあるため,これらの反応は適切な場所において迅速また正確に起こる必要があり,通常はこれらの機能は厳密に制御されている.貪食細胞ではNADPHオキシダーゼという酵素がこの活性酸素種の産生にかかわっている.
活性酸素種の不適切な産生はさまざまな疾患や炎症の病態において問題となっている.たとえば,NADPHオキシダーゼの構成タンパク質をコードする遺伝子に異常をもつ慢性肉芽腫症の患者の好中球は,活性酸素種を産生できないため病原体の殺傷能がきわめて低く患者は重篤な感染症をくり返す.一方,いくつかの慢性の炎症疾患では好中球の機能の亢進により組織の損傷やDNA損傷,アポトーシスや好中球の減少をひき起こすことがある2).
この研究では,Tecファミリーに属するチロシンキナーゼのひとつBtk(Burton’s tyrosine kinase)をコードするBTK遺伝子の異常により起こる,X連鎖無γグロブリン血症(X-linked agammaglobulinemia:XLA)の患者において好中球の減少が起こるという現象に着目した.X連鎖無γグロブリン血症の患者の11~30%では初発時あるいは細菌感染症にともない好中球の減少を呈し,場合によっては生命の危険になることが知られている.しかし,その原因は長らく不明であった3,4).
BtkはT細胞には発現を認めないが,形質細胞を除くB細胞,単球,顆粒球,血小板など血球系細胞の全般に発現している.とくに,プレB細胞受容体における機能が重要で,B細胞の初期分化に重要な役割をはたしている.X連鎖無γグロブリン血症の患者はBtkの機能不全のため成熟B細胞が欠損し,抗体の産生がないため重篤な細菌感染症にくり返しかかりやすく,肺炎,気管支炎,中耳炎,副鼻腔炎,皮膚化膿症,髄膜炎,敗血症などを起こす5,6).
近年,BtkはB細胞以外でも自然免疫系のシグナル伝達,あるいは,貪食細胞系のシグナル伝達に重要であることがわかってきた7).しかし,ヒトにおける検討は少なく,また,好中球におけるシグナル伝達については不明な点が多い.この研究では,X連鎖無γグロブリン血症の患者における好中球の減少の分子機構を明らかにすることを目的とした.解析を進めるなかで,Btkはヒトの好中球において活性酸素の産生やアポトーシスを制御する重要なタンパク質であることが明らかになり,その分子機構を深く掘り下げた.
Btkの自然変異体であるXIDマウスの単球や好中球は刺激ののちの活性酸素種の産生が低いとの報告がある8).そこでまず,X連鎖無γグロブリン血症患者の末梢血よりBtkを欠損した好中球を分離し,ホルボールエステルにより刺激して,活性酸素種の産生について検討した.ヒトの好中球ではマウスでの報告とは異なり,活性酸素種の過剰な産生が起こっていた.好中球における活性酸素種の産生にはプライミングと活性化という2つの経路が必要である.そこで,リポ多糖などToll様受容体のアゴニストあるいはTNF(tumor necrosis factor,腫瘍壊死因子)によるプライミングの刺激ののち,合成ペプチドfMLPにより活性化の刺激をあたえると,Btkを欠損した好中球では活性酸素種の過剰な産生が観察された.このような活性酸素種の過剰な産生は好酸球においても認められたが,単球における活性酸素種の産生は健常人とほぼ同じ程度であった.したがって,これらはおそらく顆粒球系の細胞に特徴的な現象ではないかと思われた.
活性酸素種の過剰な産生はアポトーシスをひき起こす可能性がある2).そこで,活性化刺激ののちのアポトーシスについて検討したところ,Btkを欠損した好中球ではホルボールエステルやTNF + fMLPなどさまざまな刺激により誘導される細胞死が亢進していた.ミトコンドリア膜電位の変動や活性型カスパーゼ3の検出などから,この細胞死はアポトーシスによるものであることがわかった.これらの現象は,健常人の好中球をBtkの阻害剤と前培養しても同様に観察され,また,膜透過性のペプチドであるHph-1を用いたタンパク質導入系によりBtkを欠損した好中球に正常なBtkを発現させると,活性酸素種の産生およびアポトーシスは正常な好中球のレベルにまで回復した.したがって,観察された活性酸素種の産生の亢進およびそれにともなうアポトーシスの亢進はBtkの欠損によるものであり,おそらく,X連鎖無γグロブリン血症の患者で観察される好中球の減少も好中球の分化障害によってではなく,末梢の好中球でのBtkの欠損そのものが関与していることが示唆された.
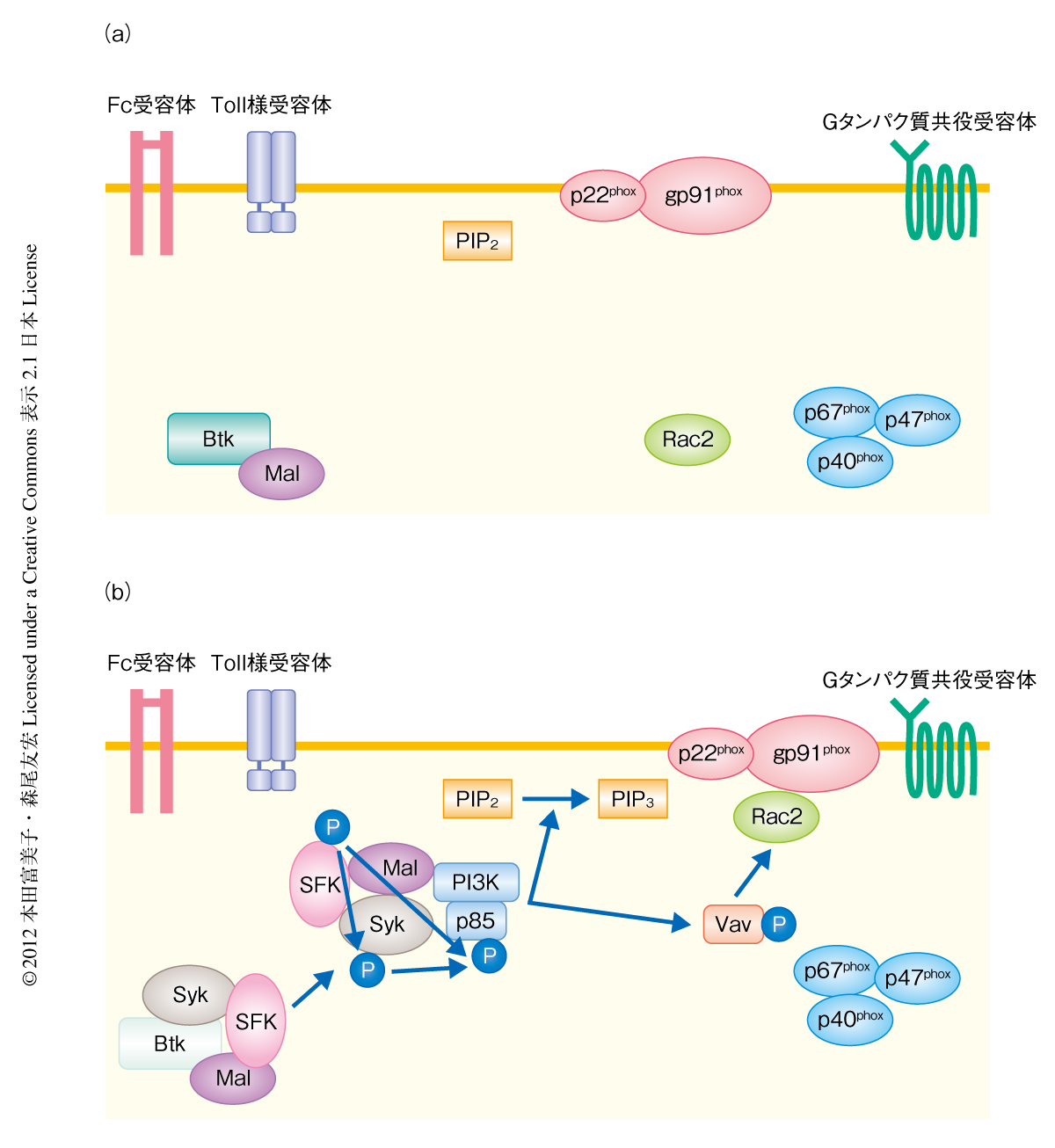
Btkが欠損するとどうして活性酸素種の産生が亢進するのだろうか? 好中球による活性酸素種の産生には,シトクロムb558とよばれるgp91phoxとp22phoxからなる“細胞膜成分”と,p47phox,p40phox,p67phoxと低分子タンパク質Rac2の4つの“細胞質成分”から構成されるNADPHオキシダーゼの活性化が必要である.NADPHオキシダーゼは細胞質成分すべてが細胞膜に移行しそこで細胞膜成分と会合することにより活性化され,その結果,活性酸素種を産生する9).その過程で,細胞質成分はチロシンキナーゼなどによりリン酸化をうける.そこでまず,NADPHオキシダーゼ構成タンパク質の細胞質や細胞膜における分布および発現,および,細胞質成分のリン酸化について調べた.その結果,Btkを欠損した好中球では静止期にてすでにgp91phoxとRac2が細胞膜において共局在しており,一方,細胞質成分であるp47phoxやp40phoxのリン酸化,p67phoxを含めたその局在は刺激の前と後のいずれにおいても正常な好中球とほぼ同じ程度であった.
なぜBtkが欠損すると未刺激の状態でもRac2が細胞膜に移行しているかについて検討した.NADPHオキシダーゼのプライミングおよび活性化の段階において,さまざまなチロシンキナーゼが関与していることが知られている.そこで,Btkを欠損した好中球における細胞内でのチロシンリン酸化の状態を検討すると,Btkを欠損した好中球では未刺激の状態(定常状態)においていくつかのタンパク質の過剰なリン酸化を認めた.単球ではこのようなチロシンリン酸化はまったく観察されなかった.したがって,活性酸素種の過剰な産生と同様に,この現象は顆粒球(好中球)に特徴的であることが示唆された.これらBtkを欠損した好中球で認められた過剰にリン酸化されたタンパク質は,PI3K(ホスファチジルイノシトール-3-キナーゼ)p85調節サブユニットや,Vav,Lyn,Syk,c-Src,Fakなどのキナーゼであることが証明された.さらに,Btkを欠損した好中球では定常状態でもホスファチジルイノシトール3,4,5-トリスリン酸の産生が増加していたことから,X連鎖無γグロブリン血症の患者の好中球ではクラス1AのPI3Kが機能的に活性化しており,それによりその下流のRac2が細胞膜成分に結合できるようになっていることが示された.また,Btkを欠損した好中球はfMLPのみの刺激でも顕著な活性酸素種の過剰産生を認め,これはPI3Kの阻害剤を処理すると正常な好中球のレベルにまで回復することがわかった.
これらの結果より,Btkを欠損した好中球ではNADPHオキシダーゼが無刺激の状態においてすでにプライミングの状態にあり,それにはPI3Kの活性化が強く関与していることが明らかになった.
Btkの欠損によりなぜPI3Kの活性化が起こるのだろうか? BtkとPI3Kとをつなぐタンパク質としてMal(MyD88-adaptor-like)に着目した.MalはToll様受容体の下流ではたらきToll様受容体への刺激ののちBtkによりリン酸化をうけることや,Btkと125番目のPro残基において結合していることが知られている7).また,単球系の細胞株においてMalは刺激ののち細胞膜に局在し,PI3Kの細胞膜への移行と活性化を促進するはたらきがあるとの報告がある10).しかしこれまで,ヒトの好中球におけるMalとBtk,また,MalとPI3Kとの関係についてはほとんど知られていない.そこでまず,健常人の好中球におけるMalとBtkの局在や会合について共免疫沈降法や免疫染色法により調べたところ,予想外にも,正常な好中球では未刺激の状態においてBtkはMalと細胞質において会合していることがわかった.一方,Btkを欠損した好中球ではMalはすでに細胞膜に局在しており,また,細胞膜においてMalとPI3Kとの会合が認められた.健常人においてMalは細胞質にあり,刺激をうけてはじめてMalとPI3Kとが細胞膜に共局在するようになった.これらの結果は,BtkがMalを細胞質にとどめるはたらきをしていることを示唆した.BtkがないとMalは細胞膜へと移行しやすくなり,細胞膜においてPI3Kとの会合をひき起こすようであった.
MalがBtkと会合する様式は知られているが,Btkのどの領域がMalと結合しているのかは明らかになっていない.そこで,Btkのそれぞれの機能ドメイン(PHドメイン,THドメイン,SH3ドメイン,SH2ドメイン,キナーゼドメイン)を欠損させた変異体を組換えタンパク質として用意し,Malとの会合に必要な領域について検討した.その結果,Malとの結合にはBtkのPHドメインとキナーゼドメインとが必要であり,MalはBtkの構造を認識しているであろうことがわかった.
さらに,PI3Kの活性化にはSrcファミリーキナーゼおよびSykの活性化が関与していることがそれぞれの阻害剤を用いた実験から示された.また,SrcファミリーキナーゼはPI3Kのリン酸化やMalの細胞膜へのターゲティングにも重要であることが明らかになった.また,Srcファミリーキナーゼ,Syk,Btk,Malはヒトの正常な好中球において通常は細胞質に共局在していた.BtkがないとSrcファミリーキナーゼの活性化によりMalは細胞膜へと移行しPI3Kを活性化する.なぜBtkが欠損するとSrcファミリーキナーゼが活性化するのかは不明で,いくつかの仮説を検証しているところである.BtkがCskやCsk結合タンパク質,ホスファターゼなどの負の制御タンパク質の活性を制御している可能性についてはいまのところ否定的である.一方,SrcファミリーキナーゼはMalと会合しているが,Btkの非存在下における会合がSrcファミリーキナーゼを活性化することも考えられ今後の検討課題である.また,正常な好中球においてSrcファミリーキナーゼが好中球のプライミングにどのような役割をはたしているかについても検討していく必要がある.
この研究により,Btkは細胞質においてMalと結合しこれを細胞質にとどめることで,好中球の反応を制御するゲートキーパーの役割をはたしていることが示された.また,X連鎖無γグロブリン血症の患者において好中球の減少の起こる分子機構は,過剰な活性酸素種を介した好中球のアポトーシスによるものであることも明らかにされた(図1).X連鎖無γグロブリン血症の患者に対してはγグロブリンの補充により好中球の減少が起こりにくくなるが,これは感染症の頻度が低下するためと考えられた.
好中球の活性化により活性酸素種が過剰に産生され臓器が損傷をうけるさまざまな疾患が知られている.活性酸素種の過剰な産生は動脈硬化や老化にも関与する.また,重症な感染症では好中球がつぎつぎとアポトーシスを起こし肺などの臓器を障害するとともに,好中球の減少により感染症の制御がさらにむずかしくなることがある.今回の研究成果を生かすことで,好中球からの活性酸素種の産生の制御が可能になるとともに,さまざまな疾患において問題となる好中球の過剰な反応を抑えることが可能になることが期待される.
Btkの欠損においてはB細胞の初期分化が障害されていて,BtkはとくにプレB細胞受容体のシグナル伝達において重要とされているが,プレB細胞の段階で分化のとまる詳細な分子機構はいまだ不明である.たとえば,プレB細胞では受容体の発現自体がシグナル伝達につながるとされている.もしかすると,特定の分化の段階ではBtkが欠損すると好中球で観察されたように活性化の刺激ののちのアポトーシスが亢進するのかもしれない.
略歴:東京医科歯科大学大学院博士後期課程 在学中.
研究テーマ:先天性免疫不全症候群の病態の解析および治療法の探索.
抱負:免疫不全症候群の病態の解析をとおして,新たな免疫の機能を知り,患者さんの治療に貢献していきたい.
森尾 友宏(Tomohiro Morio)
東京医科歯科大学大学院医歯学総合研究科 准教授.
研究室URL:http://www.tmd.ac.jp/med/ped/
© 2012 本田富美子・森尾友宏 Licensed under CC 表示 2.1 日本
(東京医科歯科大学大学院医歯学総合研究科 発生発達病態学)
email:本田富美子,森尾友宏
DOI: 10.7875/first.author.2012.036
The kinase Btk negatively regulates the production of reactive oxygen species and stimulation-induced apoptosis in human neutrophils.
Fumiko Honda, Hirotsugu Kano, Hirokazu Kanegane, Shigeaki Nonoyama, Eun-Sung Kim, Sang-Kyou Lee, Masatoshi Takagi, Shuki Mizutani, Tomohiro Morio
Nature Immunology, 13, 369-378 (2012)
要 約
X連鎖無γグロブリン血症はBTK遺伝子の異常によりB細胞の欠損する免疫不全症である.この疾患では軽い細菌感染症によっても好中球の減少が起こり,場合によっては生命の危険になることが知られている.この研究において,筆者らは,BTK遺伝子によりコードされているチロシンキナーゼBtkが好中球において特別のはたらきをしていて,刺激の入るまえにはMalが細胞膜へと移動するのを抑え,好中球による活性酸素種の産生におけるプライミングの段階を微調整し,軽い刺激に反応して活性酸素種を過剰に産生しないよう制御していることを明らかにした.また,好中球においてMalを制御する詳細なシグナル伝達機構をはじめて示した.さらに,好中球にBtkを効率よく導入したりその機能を抑えたりすることで,好中球からの活性酸素種の産生やアポトーシスを制御できることがわかった.
はじめに
好中球は微生物の感染の初期に動員され貪食や殺傷を行う,生体防御において重要な細胞である1).取り込んだ病原体はさまざまな機構により殺傷されるが,その中心的な役割をするのが活性酸素種である.活性酸素種は細胞毒性をもち自らの細胞を傷つけるなどのおそれがあるため,これらの反応は適切な場所において迅速また正確に起こる必要があり,通常はこれらの機能は厳密に制御されている.貪食細胞ではNADPHオキシダーゼという酵素がこの活性酸素種の産生にかかわっている.
活性酸素種の不適切な産生はさまざまな疾患や炎症の病態において問題となっている.たとえば,NADPHオキシダーゼの構成タンパク質をコードする遺伝子に異常をもつ慢性肉芽腫症の患者の好中球は,活性酸素種を産生できないため病原体の殺傷能がきわめて低く患者は重篤な感染症をくり返す.一方,いくつかの慢性の炎症疾患では好中球の機能の亢進により組織の損傷やDNA損傷,アポトーシスや好中球の減少をひき起こすことがある2).
この研究では,Tecファミリーに属するチロシンキナーゼのひとつBtk(Burton’s tyrosine kinase)をコードするBTK遺伝子の異常により起こる,X連鎖無γグロブリン血症(X-linked agammaglobulinemia:XLA)の患者において好中球の減少が起こるという現象に着目した.X連鎖無γグロブリン血症の患者の11~30%では初発時あるいは細菌感染症にともない好中球の減少を呈し,場合によっては生命の危険になることが知られている.しかし,その原因は長らく不明であった3,4).
BtkはT細胞には発現を認めないが,形質細胞を除くB細胞,単球,顆粒球,血小板など血球系細胞の全般に発現している.とくに,プレB細胞受容体における機能が重要で,B細胞の初期分化に重要な役割をはたしている.X連鎖無γグロブリン血症の患者はBtkの機能不全のため成熟B細胞が欠損し,抗体の産生がないため重篤な細菌感染症にくり返しかかりやすく,肺炎,気管支炎,中耳炎,副鼻腔炎,皮膚化膿症,髄膜炎,敗血症などを起こす5,6).
近年,BtkはB細胞以外でも自然免疫系のシグナル伝達,あるいは,貪食細胞系のシグナル伝達に重要であることがわかってきた7).しかし,ヒトにおける検討は少なく,また,好中球におけるシグナル伝達については不明な点が多い.この研究では,X連鎖無γグロブリン血症の患者における好中球の減少の分子機構を明らかにすることを目的とした.解析を進めるなかで,Btkはヒトの好中球において活性酸素の産生やアポトーシスを制御する重要なタンパク質であることが明らかになり,その分子機構を深く掘り下げた.
1.Btkを欠損した好中球では活性酸素種の過剰な産生により早期にアポトーシスを起こす
Btkの自然変異体であるXIDマウスの単球や好中球は刺激ののちの活性酸素種の産生が低いとの報告がある8).そこでまず,X連鎖無γグロブリン血症患者の末梢血よりBtkを欠損した好中球を分離し,ホルボールエステルにより刺激して,活性酸素種の産生について検討した.ヒトの好中球ではマウスでの報告とは異なり,活性酸素種の過剰な産生が起こっていた.好中球における活性酸素種の産生にはプライミングと活性化という2つの経路が必要である.そこで,リポ多糖などToll様受容体のアゴニストあるいはTNF(tumor necrosis factor,腫瘍壊死因子)によるプライミングの刺激ののち,合成ペプチドfMLPにより活性化の刺激をあたえると,Btkを欠損した好中球では活性酸素種の過剰な産生が観察された.このような活性酸素種の過剰な産生は好酸球においても認められたが,単球における活性酸素種の産生は健常人とほぼ同じ程度であった.したがって,これらはおそらく顆粒球系の細胞に特徴的な現象ではないかと思われた.
活性酸素種の過剰な産生はアポトーシスをひき起こす可能性がある2).そこで,活性化刺激ののちのアポトーシスについて検討したところ,Btkを欠損した好中球ではホルボールエステルやTNF + fMLPなどさまざまな刺激により誘導される細胞死が亢進していた.ミトコンドリア膜電位の変動や活性型カスパーゼ3の検出などから,この細胞死はアポトーシスによるものであることがわかった.これらの現象は,健常人の好中球をBtkの阻害剤と前培養しても同様に観察され,また,膜透過性のペプチドであるHph-1を用いたタンパク質導入系によりBtkを欠損した好中球に正常なBtkを発現させると,活性酸素種の産生およびアポトーシスは正常な好中球のレベルにまで回復した.したがって,観察された活性酸素種の産生の亢進およびそれにともなうアポトーシスの亢進はBtkの欠損によるものであり,おそらく,X連鎖無γグロブリン血症の患者で観察される好中球の減少も好中球の分化障害によってではなく,末梢の好中球でのBtkの欠損そのものが関与していることが示唆された.
2.Btkを欠損した好中球は休止期にすでにプライミングの状態にある
Btkが欠損するとどうして活性酸素種の産生が亢進するのだろうか? 好中球による活性酸素種の産生には,シトクロムb558とよばれるgp91phoxとp22phoxからなる“細胞膜成分”と,p47phox,p40phox,p67phoxと低分子タンパク質Rac2の4つの“細胞質成分”から構成されるNADPHオキシダーゼの活性化が必要である.NADPHオキシダーゼは細胞質成分すべてが細胞膜に移行しそこで細胞膜成分と会合することにより活性化され,その結果,活性酸素種を産生する9).その過程で,細胞質成分はチロシンキナーゼなどによりリン酸化をうける.そこでまず,NADPHオキシダーゼ構成タンパク質の細胞質や細胞膜における分布および発現,および,細胞質成分のリン酸化について調べた.その結果,Btkを欠損した好中球では静止期にてすでにgp91phoxとRac2が細胞膜において共局在しており,一方,細胞質成分であるp47phoxやp40phoxのリン酸化,p67phoxを含めたその局在は刺激の前と後のいずれにおいても正常な好中球とほぼ同じ程度であった.
なぜBtkが欠損すると未刺激の状態でもRac2が細胞膜に移行しているかについて検討した.NADPHオキシダーゼのプライミングおよび活性化の段階において,さまざまなチロシンキナーゼが関与していることが知られている.そこで,Btkを欠損した好中球における細胞内でのチロシンリン酸化の状態を検討すると,Btkを欠損した好中球では未刺激の状態(定常状態)においていくつかのタンパク質の過剰なリン酸化を認めた.単球ではこのようなチロシンリン酸化はまったく観察されなかった.したがって,活性酸素種の過剰な産生と同様に,この現象は顆粒球(好中球)に特徴的であることが示唆された.これらBtkを欠損した好中球で認められた過剰にリン酸化されたタンパク質は,PI3K(ホスファチジルイノシトール-3-キナーゼ)p85調節サブユニットや,Vav,Lyn,Syk,c-Src,Fakなどのキナーゼであることが証明された.さらに,Btkを欠損した好中球では定常状態でもホスファチジルイノシトール3,4,5-トリスリン酸の産生が増加していたことから,X連鎖無γグロブリン血症の患者の好中球ではクラス1AのPI3Kが機能的に活性化しており,それによりその下流のRac2が細胞膜成分に結合できるようになっていることが示された.また,Btkを欠損した好中球はfMLPのみの刺激でも顕著な活性酸素種の過剰産生を認め,これはPI3Kの阻害剤を処理すると正常な好中球のレベルにまで回復することがわかった.
これらの結果より,Btkを欠損した好中球ではNADPHオキシダーゼが無刺激の状態においてすでにプライミングの状態にあり,それにはPI3Kの活性化が強く関与していることが明らかになった.
3.BtkはMalを細胞質にとどめることで活性酸素種の産生を微調整している
Btkの欠損によりなぜPI3Kの活性化が起こるのだろうか? BtkとPI3Kとをつなぐタンパク質としてMal(MyD88-adaptor-like)に着目した.MalはToll様受容体の下流ではたらきToll様受容体への刺激ののちBtkによりリン酸化をうけることや,Btkと125番目のPro残基において結合していることが知られている7).また,単球系の細胞株においてMalは刺激ののち細胞膜に局在し,PI3Kの細胞膜への移行と活性化を促進するはたらきがあるとの報告がある10).しかしこれまで,ヒトの好中球におけるMalとBtk,また,MalとPI3Kとの関係についてはほとんど知られていない.そこでまず,健常人の好中球におけるMalとBtkの局在や会合について共免疫沈降法や免疫染色法により調べたところ,予想外にも,正常な好中球では未刺激の状態においてBtkはMalと細胞質において会合していることがわかった.一方,Btkを欠損した好中球ではMalはすでに細胞膜に局在しており,また,細胞膜においてMalとPI3Kとの会合が認められた.健常人においてMalは細胞質にあり,刺激をうけてはじめてMalとPI3Kとが細胞膜に共局在するようになった.これらの結果は,BtkがMalを細胞質にとどめるはたらきをしていることを示唆した.BtkがないとMalは細胞膜へと移行しやすくなり,細胞膜においてPI3Kとの会合をひき起こすようであった.
MalがBtkと会合する様式は知られているが,Btkのどの領域がMalと結合しているのかは明らかになっていない.そこで,Btkのそれぞれの機能ドメイン(PHドメイン,THドメイン,SH3ドメイン,SH2ドメイン,キナーゼドメイン)を欠損させた変異体を組換えタンパク質として用意し,Malとの会合に必要な領域について検討した.その結果,Malとの結合にはBtkのPHドメインとキナーゼドメインとが必要であり,MalはBtkの構造を認識しているであろうことがわかった.
さらに,PI3Kの活性化にはSrcファミリーキナーゼおよびSykの活性化が関与していることがそれぞれの阻害剤を用いた実験から示された.また,SrcファミリーキナーゼはPI3Kのリン酸化やMalの細胞膜へのターゲティングにも重要であることが明らかになった.また,Srcファミリーキナーゼ,Syk,Btk,Malはヒトの正常な好中球において通常は細胞質に共局在していた.BtkがないとSrcファミリーキナーゼの活性化によりMalは細胞膜へと移行しPI3Kを活性化する.なぜBtkが欠損するとSrcファミリーキナーゼが活性化するのかは不明で,いくつかの仮説を検証しているところである.BtkがCskやCsk結合タンパク質,ホスファターゼなどの負の制御タンパク質の活性を制御している可能性についてはいまのところ否定的である.一方,SrcファミリーキナーゼはMalと会合しているが,Btkの非存在下における会合がSrcファミリーキナーゼを活性化することも考えられ今後の検討課題である.また,正常な好中球においてSrcファミリーキナーゼが好中球のプライミングにどのような役割をはたしているかについても検討していく必要がある.
おわりに
この研究により,Btkは細胞質においてMalと結合しこれを細胞質にとどめることで,好中球の反応を制御するゲートキーパーの役割をはたしていることが示された.また,X連鎖無γグロブリン血症の患者において好中球の減少の起こる分子機構は,過剰な活性酸素種を介した好中球のアポトーシスによるものであることも明らかにされた(図1).X連鎖無γグロブリン血症の患者に対してはγグロブリンの補充により好中球の減少が起こりにくくなるが,これは感染症の頻度が低下するためと考えられた.
好中球の活性化により活性酸素種が過剰に産生され臓器が損傷をうけるさまざまな疾患が知られている.活性酸素種の過剰な産生は動脈硬化や老化にも関与する.また,重症な感染症では好中球がつぎつぎとアポトーシスを起こし肺などの臓器を障害するとともに,好中球の減少により感染症の制御がさらにむずかしくなることがある.今回の研究成果を生かすことで,好中球からの活性酸素種の産生の制御が可能になるとともに,さまざまな疾患において問題となる好中球の過剰な反応を抑えることが可能になることが期待される.
Btkの欠損においてはB細胞の初期分化が障害されていて,BtkはとくにプレB細胞受容体のシグナル伝達において重要とされているが,プレB細胞の段階で分化のとまる詳細な分子機構はいまだ不明である.たとえば,プレB細胞では受容体の発現自体がシグナル伝達につながるとされている.もしかすると,特定の分化の段階ではBtkが欠損すると好中球で観察されたように活性化の刺激ののちのアポトーシスが亢進するのかもしれない.
文 献
- Flannagan, R. S., Cosio, G. & Grinstein, S.: Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat. Rev. Microbiol., 7, 355-366 (2009)[PubMed]
- Finkel, T.: Radical medicine: treating ageing to cure disease. Nat. Rev. Mol. Cell Biol., 6, 971-976 (2005)[PubMed]
- Winkelstein, J. A., Marino. M. C., Lederman, H. M. et al.: X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine, 85, 193-202 (2006)[PubMed]
- Conley, M. E., Broides, A., Hernandez-Trujillo, V. et al.: Genetic analysis of patients with defects in early B-cell development. Immunol. Rev., 203, 216-234 (2005)[PubMed]
- Mohamed, A. J., Yu, L., Backesjo, C. M. et al.: Bruton's tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol. Rev., 228, 58-73 (2009)[PubMed]
- Doyle, S. L., Jefferies, C. A., Feighery, C. et al.: Signaling by Toll-like receptors 8 and 9 requires Bruton's tyrosine kinase. J. Biol. Chem., 282, 36953-36960 (2007)[PubMed]
- O'Neill, L. A. & Bowie, A. G.: The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol., 7, 353-364 (2007)[PubMed]
- Mangla, A., Khare, A., Vineeth, V. et al.: Pleiotropic consequences of Bruton tyrosine kinase deficiency in myeloid lineages lead to poor inflammatory responses. Blood, 104, 1191-1197 (2004)[PubMed]
- Babior, B. M.: NADPH oxidase. Curr. Opin. Immunol., 16, 42-47 (2004)[PubMed]
- Santos-Sierra, S., Deshmukh, S. D., Kalnitski, J. et al.: Mal connects TLR2 to PI3Kinase activation and phagocyte polarization. EMBO J., 28, 2018-2027 (2009)[PubMed]
著者プロフィール
略歴:東京医科歯科大学大学院博士後期課程 在学中.
研究テーマ:先天性免疫不全症候群の病態の解析および治療法の探索.
抱負:免疫不全症候群の病態の解析をとおして,新たな免疫の機能を知り,患者さんの治療に貢献していきたい.
森尾 友宏(Tomohiro Morio)
東京医科歯科大学大学院医歯学総合研究科 准教授.
研究室URL:http://www.tmd.ac.jp/med/ped/
© 2012 本田富美子・森尾友宏 Licensed under CC 表示 2.1 日本
