PPARα-Sirt1複合体のERR応答配列を介した転写抑制による心機能の制御
岡 新一・佐渡島純一
(米国Medicine and Dentistry of New Jersey大学,Department of Cell Biology and Molecular Medicine)
email:岡 新一,佐渡島純一
DOI: 10.7875/first.author.2011.175
PPARα-Sirt1 complex mediates cardiac hypertrophy and failure through suppression of the ERR transcriptional pathway.
Shinichi Oka, Ralph Alcendor, Peiyong Zhai, Ji Yeon Park, Dan Shao, Jaeyeaon Cho, Takanobu Yamamoto, Bin Tian, Junichi Sadoshima
Cell Metabolism, 14, 598-611 (2011)
ミトコンドリアによるエネルギーの産生は心機能の維持に必須である.心不全の際にはミトコンドリアにおいてエネルギーの産生にかかわる遺伝子の発現低下が観察されるが,その分子機構は不明である.この研究において,筆者らは,圧力負荷による心不全誘導モデルマウスにおけるミトコンドリア遺伝子の発現低下に,核内受容体であるPPARαとNAD依存性タンパク質脱アセチル化酵素であるSirt1の関与することを見い出した.PPARαはSirt1と複合体を形成しERR応答配列に結合することにより遺伝子発現を抑制した.ERR応答配列は多くのミトコンドリア遺伝子のプロモーター領域に存在するため,この機構によりミトコンドリア遺伝子の発現抑制が誘導されるものと考えられた.また,このPPARα-Sirt1複合体によるミトコンドリア遺伝子の発現抑制は生理的な絶食の際にも起こることが示された.すなわち,本来,生体に備わった絶食応答が,心不全の際にはミトコンドリア遺伝子の発現低下を誘導していることが示唆された.
心臓はつねに収縮し全身に血液を送り出しているため,消費にみあった多くのエネルギーの産生を必要とする臓器である.このため,心筋細胞には大量のミトコンドリアが存在しエネルギーの産生を担っている.ヒトにおいて,とくに先進国では,心不全は主要な死亡原因のひとつである.心不全の際の心筋細胞ではミトコンドリアにおけるエネルギーの産生にかかわる遺伝子の発現低下が観察される1).ミトコンドリアの機能の不全はエネルギー産生能の低下や細胞死による心筋細胞の脱落を誘導し,心機能の不全を助長するものと考えられている.
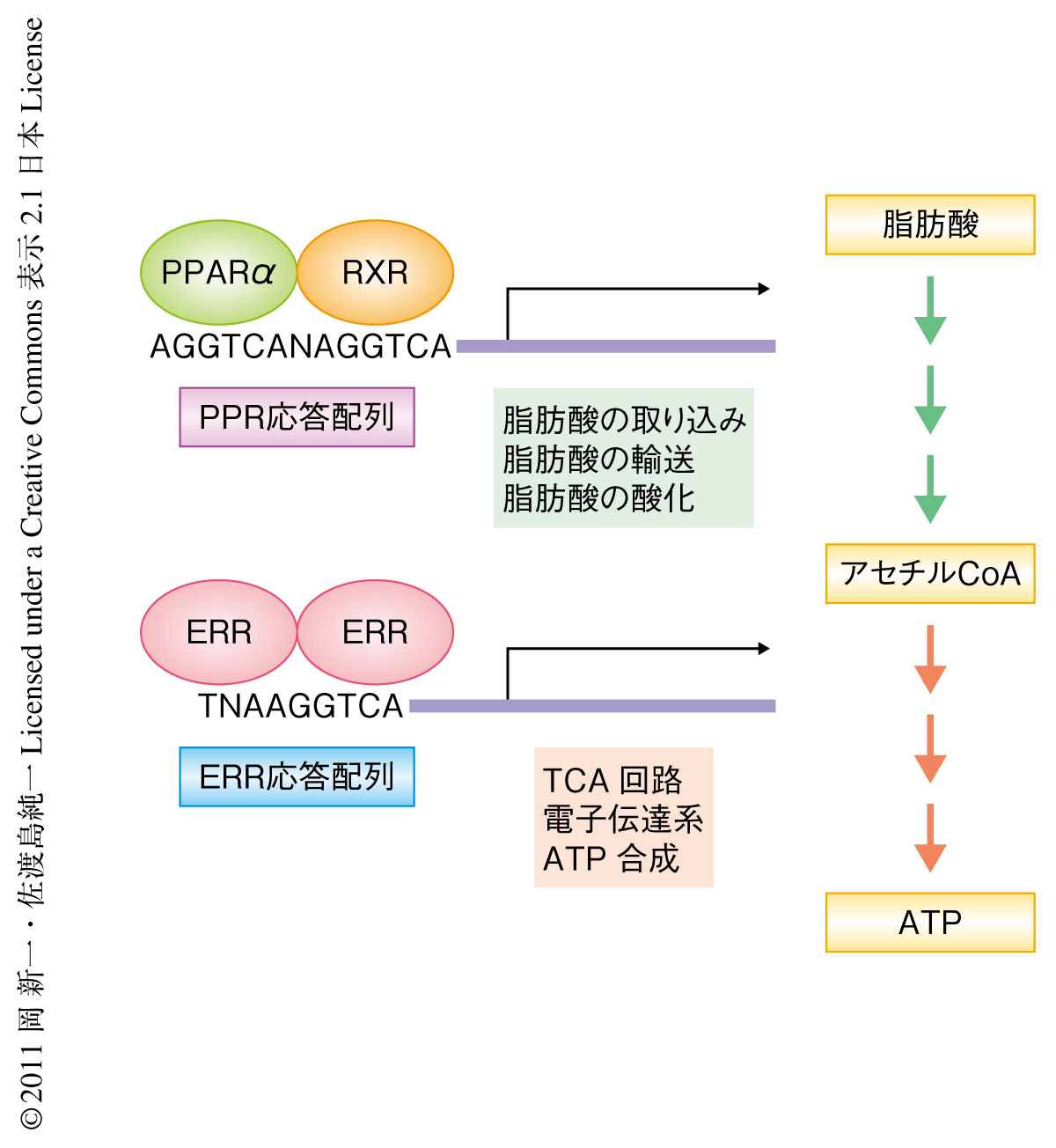
核内受容体ファミリーは発生,免疫応答やエネルギー代謝など,さまざまな生体機能を担う転写因子から構成されている.核内受容体のDNA結合配列はAGGTCA配列やAGAACA配列など共通の6塩基単位の配列を2つ組み合わせた配列,または,この配列をひとつだけ含む配列により構成されている2).ERR(estrogen related receptor,エストロゲン関連受容体)は核内受容体ファミリーのひとつであるが,ERR応答配列はAGGTCA配列をひとつだけ含むTNAAGGTCA(Nは任意の塩基)というコンセンサス配列からなり,TCA回路や呼吸鎖などミトコンドリアにおけるエネルギーの産生を担うタンパク質をコードする多くの遺伝子のプロモーター領域に存在する.そのため,ERRはエネルギーの産生に重要な転写因子と考えられている3)(図1).PPAR(peroxisome proliferator-activated receptor,ペルオキシソーム増殖因子活性化受容体)も核内受容体ファミリーのひとつであり,α,β/δ,γ,の3つのアイソフォームが存在する.PPARは別の核内受容体ファミリーのひとつであるRXR(retinoid X receptor,レチノイドX受容体)とヘテロ二量体を形成し転写の活性化能をもつようになる.PPAR応答配列は2つのAGGTCA配列が任意の1塩基をはさみ同じ方向に並ぶAGGTCANAGGTCAというコンセンサス配列からなる.PPARは5’側にあるAGGTCA配列,RXRは3’側にあるAGGTCA配列へ結合する.PPARαは脂肪酸の代謝にかかわる酵素の遺伝子の転写を活性化し,とりわけ,絶食の際に活性化する.体外からの栄養補給が絶たれたときには体内に蓄えた脂肪分がおもな栄養素となるためである(図1).
この研究では,心不全の際,PPARαがERR応答配列のAGGTCA配列を認識して結合し,その標的遺伝子の発現を抑制することを見い出した.また,この遺伝子発現の抑制にはRXRを必要とせず,NAD依存性タンパク質脱アセチル化酵素であるSirt1(silent information regulator 1)が必須の役割をはたしていることを明らかにした.
まず,免疫沈降法により内在性のPPARαとSirt1との結合を心筋初代培養細胞で確認し,PPARαとSirt1とが複合体を形成することを示した.そののち,大動脈狭窄術による心不全誘導モデルマウスを用いてPPARαおよびSirt1の役割を検討した.大動脈は心臓から全身に血液を送り出す直近の血管であり,横行大動脈を外科的に狭窄することにより左心室へ過剰の圧力負荷をあたえることでマウスに心不全を誘導することができる.この心不全誘導モデルマウスの心臓においてPPARαとSirt1のタンパク質量は上昇し,PPARαヘテロノックアウトマウスおよびSirt1ヘテロノックアウトマウスでは野生型マウスに比べ心機能が顕著に改善していた.逆に,PPARαを心臓に過剰発現するトランスジェニックマウスとSirt1を心臓に過剰発現するトランスジェニックマウスとを掛け合わせることで作製したPPARαおよびSirt1の同時過剰発現マウスは,圧力負荷をあたえなくとも重篤な心不全におちいった.これらのことから,PPARαとSirt1はともに心不全の発症に重要なタンパク質であることが示唆された.
PPARαとSirt1による心不全の誘導機構を解析するため,PPARα過剰発現マウス,Sirt1過剰発現マウス,PPARαとSirt1の同時過剰発現マウスを用いて,マイクロアレイ解析を行った.Sirt1はPPARαの転写活性を促進することが報告されていたが,筆者らのSirt1過剰発現マウスのマイクロアレイ解析ではそのような結果は得られなかった.一方,PPARα過剰発現マウスにおいては核ゲノムにコードされる多くのミトコンドリア遺伝子の発現が減少し,PPARαとSirt1の同時過剰発現マウスではさらに減少していることが見い出された.そののち,このような発現パターンを示すミトコンドリア遺伝子をひとつひとつ文献検索したところ,それらの多くがERRの標的遺伝子であることが明らかになった.ERRはミトコンドリア遺伝子のほかにも,心臓の収縮に必須であるカルシウム制御タンパク質の遺伝子やエネルギーの産生を促進するPGC1αなどの転写因子の遺伝子の転写を活性化することが知られており,心機能にはとりわけ重要な核内受容体と考えられている4).マイクロアレイ解析においてPPARαとSirt1の同時過剰発現により発現が変化した遺伝子のなかから,ERR標的遺伝子としてすでに報告されているものやERR応答配列の存在によりERR標的遺伝子であると予想される遺伝子を抽出し,遺伝子発現プロファイルを作成した.その結果,多くのERR標的遺伝子の発現がPPARαとSirt1により協調的に抑制されていることが見い出された.
さきに述べたようにERR応答配列はTNAAGGTCAというコンセンサス配列をもち,この配列はPPAR-RXRヘテロ二量体におけるPPARのDNA結合配列であるAGGTCAを含むことから,PPARαはERR応答配列へ直接に結合し遺伝子発現を抑制することが考えられた.精製PPARαはRXRの非存在下でも単独で直接にPPAR応答配列およびERR応答配列へ結合したが,AGGTCA配列をGAATCAへと変異させた場合は結合しなかった.すなわち,PPARαは単量体として配列特異的にDNAへ結合できることが示された.クロマチン免疫沈降法により細胞におけるPPARαのDNAへの結合領域をPPARα過剰発現マウスにおいて検討したところ,PPARαはERR標的遺伝子のプロモーター領域に存在するERR応答配列の近傍に結合することが示された.また,PPARαを過剰発現させるとSirt1がERR応答配列の近傍へ結合し,PPARαによりSirt1がERR応答配列へリクルートされることが示された.ERR応答配列を用いたレポーター解析により,PPARαとSirt1はERR標的遺伝子のプロモーターの活性を協調的に抑制することが確認された.興味深いことに,このPPARαとSirt1によるERR応答配列の抑制は,野生型RXR,および,DNA結合領域を欠損した変異型RXRを過剰発現した場合にも同様に観察された.一方,PPARαによるPPAR応答配列の活性化は野生型RXRにより増強され,DNA結合領域を欠損した変異型RXRにより抑制された.すなわち,RXRはPPARαによるPPAR応答配列の活性化には必須であるが,PPARα-Sirt1複合体によるERR応答配列の抑制には要求されないことが示された.
PPARαおよびSirt1のERR応答配列の近傍への結合は,圧力負荷による心不全誘導モデルマウスの心臓においても誘導されることがクロマチン免疫沈降法により確認された.また,心不全誘導モデルマウスの心臓ではERR応答配列の近傍においてヒストンH3の9番目のリジンのアセチル化レベルが減少していた.このヒストンH3の9番目のリジンはSirt1により脱アセチル化されることから,PPARαによりリクルートされたSirt1がヒストン脱アセチル化を介して遺伝子発現を抑制していることが示唆された.PPARαヘテロノックアウトマウスおよびSirt1ヘテロノックアウトマウスでは心不全誘導モデルマウスにおけるERR標的遺伝子の発現低下が抑制されていた.これらの実験結果から,心不全誘導モデルマウスの心臓ではPPARα-Sirt1複合体がERR応答配列へ結合しミトコンドリア遺伝子の発現抑制を行っていることが示唆された.
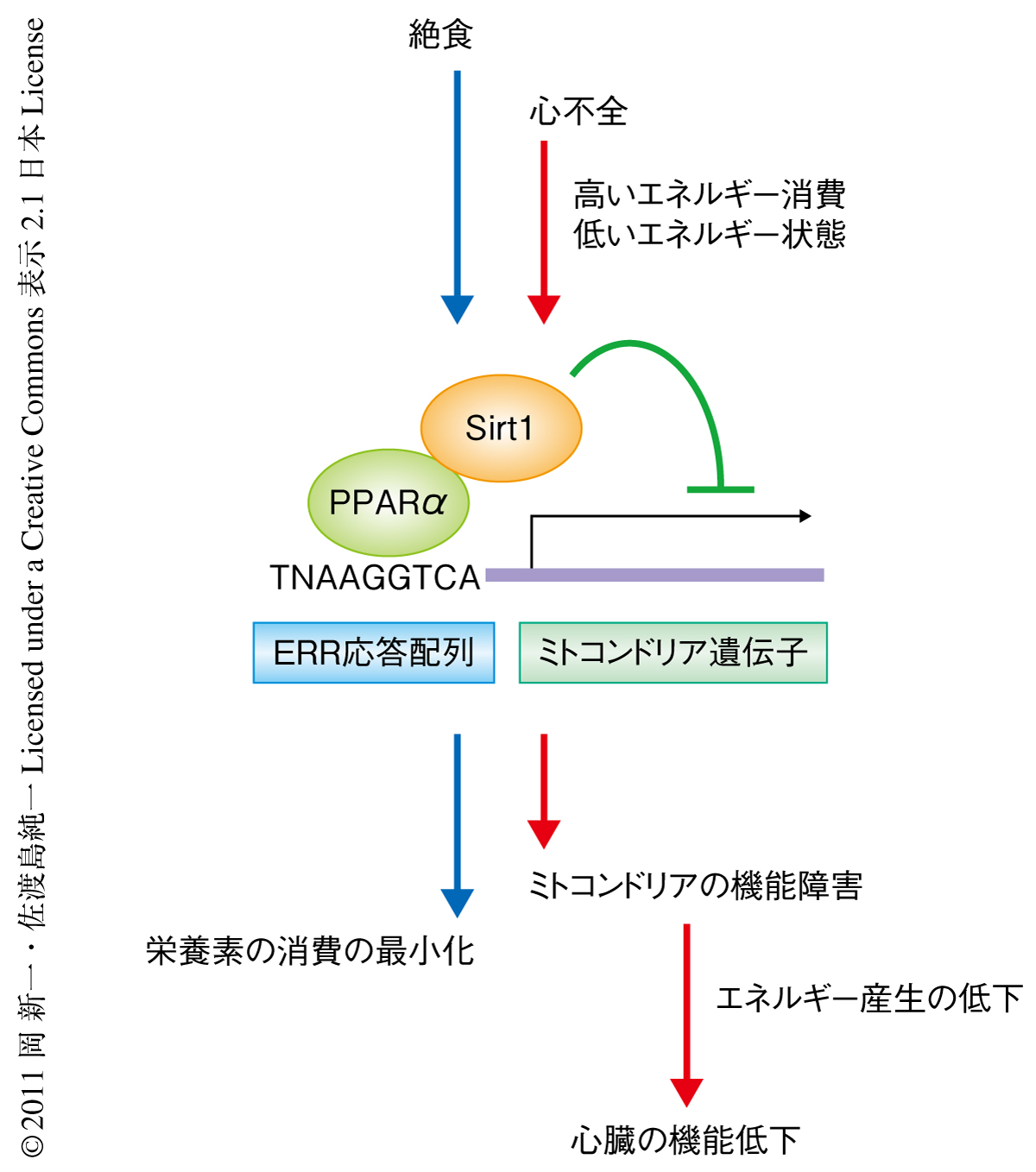
PPARαとSirt1は絶食などの低エネルギー状態への生体応答に重要なシグナル伝達タンパク質である.そこで,PPARα-Sirt1複合体によるERR標的遺伝子の発現抑制は生理的な絶食応答である可能性を検討した.絶食24時間後の野生型マウスの心臓におけるERR標的遺伝子の発現を解析したところ,その多くで発現量の低下が観察された.絶食の際のERR標的遺伝子の低下はPPARαヘテロノックアウトマウスおよびSirt1ヘテロノックアウトマウスでは抑制されており,PPARα-Sirt1複合体が絶食の際のERR標的遺伝子の発現抑制にかかわることが示唆された.絶食24時間後の野生型マウスでは心収縮能が減弱していたが,この変化はPPARαヘテロノックアウトマウスおよびSirt1ヘテロノックアウトマウスでは観察されなかった.初代培養心筋細胞はグルコースを含まない培地で培養すると細胞死が誘導されるが,PPARαおよびSirt1を過剰発現した心筋細胞ではこの細胞死が顕著に遅延した.これらの結果より,PPARα-Sirt1複合体によるERR応答配列を介した遺伝子発現の抑制は,絶食などの低エネルギー状態への重要な生理的応答であることが示唆された(図2).
この研究により,心不全の際にPPARα-Sirt1複合体が核ゲノムにコードされるミトコンドリア遺伝子の発現低下を担うことが示された.PPARαはAGGTCA様配列のくり返しからなるPPAR応答配列に結合し転写の活性化を行うことがおもな機能だと考えられている.今回,筆者らは,PPARαは2つのAGGTCA様配列をもつPPAR応答配列だけでなく,ERR応答配列など単独で存在するAGGTCA様配列にも直接に結合し転写抑制を行うことを示した.クロマチン免疫沈降-マイクロアレイ法により全ゲノムにおけるPPARαの結合領域を解析した結果,AGGTCA様配列のくり返しからなるPPAR応答配列よりも,ERR応答配列など単独で存在するAGGTCA様配列のほうが主要なPPARαの結合配列であることが明らかになっている.また,リガンド刺激によりPPARαのDNAへの結合が誘導されるが,この場合も,PPARαはリガンド刺激により発現が上昇している遺伝子だけでなく,発現が低下している遺伝子のプロモーター領域の多くに結合している5).これらのことから,単独で存在するAGGTCA様配列を介した遺伝子発現の抑制はPPARαの重要な機能のひとつであるものと考えられる.
ミトコンドリアにおけるエネルギーの産生にかかわる遺伝子の発現低下は心不全の患者において高頻度に観察される病態であるが,その意義は不明であった.とりわけ,筆者らの用いたマウス大動脈狭窄術による心不全誘導モデルマウスは心臓に高い圧力を負荷する病態モデルであり,心機能の維持のためにはより高いエネルギーの産生が要求される.しかし,心不全の際には心筋細胞はまったく逆の応答を示し,エネルギーの産生にかかわる遺伝子の発現低下を誘導する.ここでは,PPARα-Sirt1複合体によるエネルギーの産生にかかわる遺伝子の転写抑制がこの分子病態を説明する可能性を提示した.高血圧などの要因によりエネルギー消費が亢進した結果,心筋細胞が低エネルギー状態におちいると,絶食による低エネルギー状態と同じようにPPARα-Sirt1複合体が形成され,ミトコンドリアの機能を抑制するものと考えられる(図2).従属栄養生物である哺乳類は飢餓において体内に蓄えた栄養分を消費しつくしてしまうと生体機能を維持できない.このため,PPARα-Sirt1複合体による栄養素の消費抑制の機構は飢餓の際の生体機能の延命に重要であると考えられる.しかし,圧力負荷による心不全の際には栄養素の消費抑制の機構は心筋の収縮力の低下をもたらし,病態をさらに悪化させると考えられる.
略歴:2001年 姫路工業大学大学院博士後期課程 修了,同年 京都大学ウイルス研究所 博士研究員,2002年 産業技術総合研究所セルエンジニアリング研究部門 博士研究員,2005年 京都大学ウイルス研究所 博士研究員,2006年 米国Michigan大学 博士研究員,2007年 米国Medicine and Dentistry of New Jersey大学 博士研究員を経て,2008年より同 リサーチアソシエイト.
研究テーマ:心臓における代謝制御とレドックス制御.
関心事:立身出世.
佐渡島 純一(Junichi Sadoshima)
米国Medicine and Dentistry of New Jersey大学 教授.
研究室URL:http://www.sadoshimalab.org/
© 2011 岡 新一・佐渡島純一 Licensed under CC 表示 2.1 日本
(米国Medicine and Dentistry of New Jersey大学,Department of Cell Biology and Molecular Medicine)
email:岡 新一,佐渡島純一
DOI: 10.7875/first.author.2011.175
PPARα-Sirt1 complex mediates cardiac hypertrophy and failure through suppression of the ERR transcriptional pathway.
Shinichi Oka, Ralph Alcendor, Peiyong Zhai, Ji Yeon Park, Dan Shao, Jaeyeaon Cho, Takanobu Yamamoto, Bin Tian, Junichi Sadoshima
Cell Metabolism, 14, 598-611 (2011)
要 約
ミトコンドリアによるエネルギーの産生は心機能の維持に必須である.心不全の際にはミトコンドリアにおいてエネルギーの産生にかかわる遺伝子の発現低下が観察されるが,その分子機構は不明である.この研究において,筆者らは,圧力負荷による心不全誘導モデルマウスにおけるミトコンドリア遺伝子の発現低下に,核内受容体であるPPARαとNAD依存性タンパク質脱アセチル化酵素であるSirt1の関与することを見い出した.PPARαはSirt1と複合体を形成しERR応答配列に結合することにより遺伝子発現を抑制した.ERR応答配列は多くのミトコンドリア遺伝子のプロモーター領域に存在するため,この機構によりミトコンドリア遺伝子の発現抑制が誘導されるものと考えられた.また,このPPARα-Sirt1複合体によるミトコンドリア遺伝子の発現抑制は生理的な絶食の際にも起こることが示された.すなわち,本来,生体に備わった絶食応答が,心不全の際にはミトコンドリア遺伝子の発現低下を誘導していることが示唆された.
はじめに
心臓はつねに収縮し全身に血液を送り出しているため,消費にみあった多くのエネルギーの産生を必要とする臓器である.このため,心筋細胞には大量のミトコンドリアが存在しエネルギーの産生を担っている.ヒトにおいて,とくに先進国では,心不全は主要な死亡原因のひとつである.心不全の際の心筋細胞ではミトコンドリアにおけるエネルギーの産生にかかわる遺伝子の発現低下が観察される1).ミトコンドリアの機能の不全はエネルギー産生能の低下や細胞死による心筋細胞の脱落を誘導し,心機能の不全を助長するものと考えられている.
核内受容体ファミリーは発生,免疫応答やエネルギー代謝など,さまざまな生体機能を担う転写因子から構成されている.核内受容体のDNA結合配列はAGGTCA配列やAGAACA配列など共通の6塩基単位の配列を2つ組み合わせた配列,または,この配列をひとつだけ含む配列により構成されている2).ERR(estrogen related receptor,エストロゲン関連受容体)は核内受容体ファミリーのひとつであるが,ERR応答配列はAGGTCA配列をひとつだけ含むTNAAGGTCA(Nは任意の塩基)というコンセンサス配列からなり,TCA回路や呼吸鎖などミトコンドリアにおけるエネルギーの産生を担うタンパク質をコードする多くの遺伝子のプロモーター領域に存在する.そのため,ERRはエネルギーの産生に重要な転写因子と考えられている3)(図1).PPAR(peroxisome proliferator-activated receptor,ペルオキシソーム増殖因子活性化受容体)も核内受容体ファミリーのひとつであり,α,β/δ,γ,の3つのアイソフォームが存在する.PPARは別の核内受容体ファミリーのひとつであるRXR(retinoid X receptor,レチノイドX受容体)とヘテロ二量体を形成し転写の活性化能をもつようになる.PPAR応答配列は2つのAGGTCA配列が任意の1塩基をはさみ同じ方向に並ぶAGGTCANAGGTCAというコンセンサス配列からなる.PPARは5’側にあるAGGTCA配列,RXRは3’側にあるAGGTCA配列へ結合する.PPARαは脂肪酸の代謝にかかわる酵素の遺伝子の転写を活性化し,とりわけ,絶食の際に活性化する.体外からの栄養補給が絶たれたときには体内に蓄えた脂肪分がおもな栄養素となるためである(図1).
この研究では,心不全の際,PPARαがERR応答配列のAGGTCA配列を認識して結合し,その標的遺伝子の発現を抑制することを見い出した.また,この遺伝子発現の抑制にはRXRを必要とせず,NAD依存性タンパク質脱アセチル化酵素であるSirt1(silent information regulator 1)が必須の役割をはたしていることを明らかにした.
1.圧力負荷による心不全誘導モデルマウスにおけるPPARαおよびSirt1の役割
まず,免疫沈降法により内在性のPPARαとSirt1との結合を心筋初代培養細胞で確認し,PPARαとSirt1とが複合体を形成することを示した.そののち,大動脈狭窄術による心不全誘導モデルマウスを用いてPPARαおよびSirt1の役割を検討した.大動脈は心臓から全身に血液を送り出す直近の血管であり,横行大動脈を外科的に狭窄することにより左心室へ過剰の圧力負荷をあたえることでマウスに心不全を誘導することができる.この心不全誘導モデルマウスの心臓においてPPARαとSirt1のタンパク質量は上昇し,PPARαヘテロノックアウトマウスおよびSirt1ヘテロノックアウトマウスでは野生型マウスに比べ心機能が顕著に改善していた.逆に,PPARαを心臓に過剰発現するトランスジェニックマウスとSirt1を心臓に過剰発現するトランスジェニックマウスとを掛け合わせることで作製したPPARαおよびSirt1の同時過剰発現マウスは,圧力負荷をあたえなくとも重篤な心不全におちいった.これらのことから,PPARαとSirt1はともに心不全の発症に重要なタンパク質であることが示唆された.
2.PPARαとSirt1はERR標的遺伝子の発現を協調的に抑制する
PPARαとSirt1による心不全の誘導機構を解析するため,PPARα過剰発現マウス,Sirt1過剰発現マウス,PPARαとSirt1の同時過剰発現マウスを用いて,マイクロアレイ解析を行った.Sirt1はPPARαの転写活性を促進することが報告されていたが,筆者らのSirt1過剰発現マウスのマイクロアレイ解析ではそのような結果は得られなかった.一方,PPARα過剰発現マウスにおいては核ゲノムにコードされる多くのミトコンドリア遺伝子の発現が減少し,PPARαとSirt1の同時過剰発現マウスではさらに減少していることが見い出された.そののち,このような発現パターンを示すミトコンドリア遺伝子をひとつひとつ文献検索したところ,それらの多くがERRの標的遺伝子であることが明らかになった.ERRはミトコンドリア遺伝子のほかにも,心臓の収縮に必須であるカルシウム制御タンパク質の遺伝子やエネルギーの産生を促進するPGC1αなどの転写因子の遺伝子の転写を活性化することが知られており,心機能にはとりわけ重要な核内受容体と考えられている4).マイクロアレイ解析においてPPARαとSirt1の同時過剰発現により発現が変化した遺伝子のなかから,ERR標的遺伝子としてすでに報告されているものやERR応答配列の存在によりERR標的遺伝子であると予想される遺伝子を抽出し,遺伝子発現プロファイルを作成した.その結果,多くのERR標的遺伝子の発現がPPARαとSirt1により協調的に抑制されていることが見い出された.
3.PPARαはERR応答配列に結合し転写抑制を行う
さきに述べたようにERR応答配列はTNAAGGTCAというコンセンサス配列をもち,この配列はPPAR-RXRヘテロ二量体におけるPPARのDNA結合配列であるAGGTCAを含むことから,PPARαはERR応答配列へ直接に結合し遺伝子発現を抑制することが考えられた.精製PPARαはRXRの非存在下でも単独で直接にPPAR応答配列およびERR応答配列へ結合したが,AGGTCA配列をGAATCAへと変異させた場合は結合しなかった.すなわち,PPARαは単量体として配列特異的にDNAへ結合できることが示された.クロマチン免疫沈降法により細胞におけるPPARαのDNAへの結合領域をPPARα過剰発現マウスにおいて検討したところ,PPARαはERR標的遺伝子のプロモーター領域に存在するERR応答配列の近傍に結合することが示された.また,PPARαを過剰発現させるとSirt1がERR応答配列の近傍へ結合し,PPARαによりSirt1がERR応答配列へリクルートされることが示された.ERR応答配列を用いたレポーター解析により,PPARαとSirt1はERR標的遺伝子のプロモーターの活性を協調的に抑制することが確認された.興味深いことに,このPPARαとSirt1によるERR応答配列の抑制は,野生型RXR,および,DNA結合領域を欠損した変異型RXRを過剰発現した場合にも同様に観察された.一方,PPARαによるPPAR応答配列の活性化は野生型RXRにより増強され,DNA結合領域を欠損した変異型RXRにより抑制された.すなわち,RXRはPPARαによるPPAR応答配列の活性化には必須であるが,PPARα-Sirt1複合体によるERR応答配列の抑制には要求されないことが示された.
4.心不全の際のPPARα-Sirt1複合体によるERR標的遺伝子の発現抑制
PPARαおよびSirt1のERR応答配列の近傍への結合は,圧力負荷による心不全誘導モデルマウスの心臓においても誘導されることがクロマチン免疫沈降法により確認された.また,心不全誘導モデルマウスの心臓ではERR応答配列の近傍においてヒストンH3の9番目のリジンのアセチル化レベルが減少していた.このヒストンH3の9番目のリジンはSirt1により脱アセチル化されることから,PPARαによりリクルートされたSirt1がヒストン脱アセチル化を介して遺伝子発現を抑制していることが示唆された.PPARαヘテロノックアウトマウスおよびSirt1ヘテロノックアウトマウスでは心不全誘導モデルマウスにおけるERR標的遺伝子の発現低下が抑制されていた.これらの実験結果から,心不全誘導モデルマウスの心臓ではPPARα-Sirt1複合体がERR応答配列へ結合しミトコンドリア遺伝子の発現抑制を行っていることが示唆された.
5.PPARα-Sirt1複合体による遺伝子発現の抑制は絶食でも誘発される
PPARαとSirt1は絶食などの低エネルギー状態への生体応答に重要なシグナル伝達タンパク質である.そこで,PPARα-Sirt1複合体によるERR標的遺伝子の発現抑制は生理的な絶食応答である可能性を検討した.絶食24時間後の野生型マウスの心臓におけるERR標的遺伝子の発現を解析したところ,その多くで発現量の低下が観察された.絶食の際のERR標的遺伝子の低下はPPARαヘテロノックアウトマウスおよびSirt1ヘテロノックアウトマウスでは抑制されており,PPARα-Sirt1複合体が絶食の際のERR標的遺伝子の発現抑制にかかわることが示唆された.絶食24時間後の野生型マウスでは心収縮能が減弱していたが,この変化はPPARαヘテロノックアウトマウスおよびSirt1ヘテロノックアウトマウスでは観察されなかった.初代培養心筋細胞はグルコースを含まない培地で培養すると細胞死が誘導されるが,PPARαおよびSirt1を過剰発現した心筋細胞ではこの細胞死が顕著に遅延した.これらの結果より,PPARα-Sirt1複合体によるERR応答配列を介した遺伝子発現の抑制は,絶食などの低エネルギー状態への重要な生理的応答であることが示唆された(図2).
おわりに
この研究により,心不全の際にPPARα-Sirt1複合体が核ゲノムにコードされるミトコンドリア遺伝子の発現低下を担うことが示された.PPARαはAGGTCA様配列のくり返しからなるPPAR応答配列に結合し転写の活性化を行うことがおもな機能だと考えられている.今回,筆者らは,PPARαは2つのAGGTCA様配列をもつPPAR応答配列だけでなく,ERR応答配列など単独で存在するAGGTCA様配列にも直接に結合し転写抑制を行うことを示した.クロマチン免疫沈降-マイクロアレイ法により全ゲノムにおけるPPARαの結合領域を解析した結果,AGGTCA様配列のくり返しからなるPPAR応答配列よりも,ERR応答配列など単独で存在するAGGTCA様配列のほうが主要なPPARαの結合配列であることが明らかになっている.また,リガンド刺激によりPPARαのDNAへの結合が誘導されるが,この場合も,PPARαはリガンド刺激により発現が上昇している遺伝子だけでなく,発現が低下している遺伝子のプロモーター領域の多くに結合している5).これらのことから,単独で存在するAGGTCA様配列を介した遺伝子発現の抑制はPPARαの重要な機能のひとつであるものと考えられる.
ミトコンドリアにおけるエネルギーの産生にかかわる遺伝子の発現低下は心不全の患者において高頻度に観察される病態であるが,その意義は不明であった.とりわけ,筆者らの用いたマウス大動脈狭窄術による心不全誘導モデルマウスは心臓に高い圧力を負荷する病態モデルであり,心機能の維持のためにはより高いエネルギーの産生が要求される.しかし,心不全の際には心筋細胞はまったく逆の応答を示し,エネルギーの産生にかかわる遺伝子の発現低下を誘導する.ここでは,PPARα-Sirt1複合体によるエネルギーの産生にかかわる遺伝子の転写抑制がこの分子病態を説明する可能性を提示した.高血圧などの要因によりエネルギー消費が亢進した結果,心筋細胞が低エネルギー状態におちいると,絶食による低エネルギー状態と同じようにPPARα-Sirt1複合体が形成され,ミトコンドリアの機能を抑制するものと考えられる(図2).従属栄養生物である哺乳類は飢餓において体内に蓄えた栄養分を消費しつくしてしまうと生体機能を維持できない.このため,PPARα-Sirt1複合体による栄養素の消費抑制の機構は飢餓の際の生体機能の延命に重要であると考えられる.しかし,圧力負荷による心不全の際には栄養素の消費抑制の機構は心筋の収縮力の低下をもたらし,病態をさらに悪化させると考えられる.
文 献
- Karamanlidis, G., Nascimben, L., Couper, G. S. et al.: Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ. Res., 106, 1541-1548 (2010)[PubMed]
- Mangelsdorf, D. J. & Evans, R. M.: The RXR heterodimers and orphan receptors. Cell, 83, 841-850 (1995)[PubMed]
- Giguere, V.: Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocr. Rev., 29, 677-696 (2008)[PubMed]
- Dufour, C. R., Wilson, B. J., Huss, J. M. et al.: Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRα and γ. Cell Metab., 5, 345-356 (2007)[PubMed]
- van der Meer, D. L., Degenhardt, T., Vaisanen, S. et al.: Profiling of promoter occupancy by PPARα in human hepatoma cells via ChIP-chip analysis. Nucleic Acids Res., 38, 2839-2850 (2010)[PubMed]
著者プロフィール
略歴:2001年 姫路工業大学大学院博士後期課程 修了,同年 京都大学ウイルス研究所 博士研究員,2002年 産業技術総合研究所セルエンジニアリング研究部門 博士研究員,2005年 京都大学ウイルス研究所 博士研究員,2006年 米国Michigan大学 博士研究員,2007年 米国Medicine and Dentistry of New Jersey大学 博士研究員を経て,2008年より同 リサーチアソシエイト.
研究テーマ:心臓における代謝制御とレドックス制御.
関心事:立身出世.
佐渡島 純一(Junichi Sadoshima)
米国Medicine and Dentistry of New Jersey大学 教授.
研究室URL:http://www.sadoshimalab.org/
© 2011 岡 新一・佐渡島純一 Licensed under CC 表示 2.1 日本
