mTORはDEPTORの分解を誘導することにより自己の活性を増幅させる
犬塚博之・Wenyi Wei
(米国Harvard大学Medical School,Beth Israel Deaconess Medical Center,Department of Pathology)
email:犬塚博之
DOI: 10.7875/first.author.2011.174
mTOR drives its own activation via SCFβ-TrCP-dependent degradation of the mTOR inhibitor DEPTOR.
Daming Gao, Hiroyuki Inuzuka, Meng-Kwang Marcus Tan, Hidefumi Fukushima, Jason W. Locasale, Pengda Liu, Lixin Wan, Bo Zhai, Y. Rebecca Chin, Shavali Shaik, Costas A. Lyssiotis, Steven P. Gygi, Alex Toker, Lewis C. Cantley, John M. Asara, J. Wade Harper, Wenyi Wei
Molecular Cell, 44, 290-303 (2011)
mTORは機能的に異なる2種類の複合体,mTOR複合体1とmTOR複合体2を形成するが,この2つの複合体にはmTOR抑制タンパク質であるDEPTORが含まれている.低栄養状態の細胞ではDEPTORはmTORに直接結合してその活性を抑制しているが,増殖因子の刺激によりDEPTORはすみやかな分解をうけmTOR複合体の活性化を誘導する.DEPTOR分解の分子機構はこれまで不明であったが,今回,筆者らは,ユビキチンリガーゼ複合体SCFβTrCPがDEPTORの分解にかかわっていることを明らかにした.さらに,SCFβTrCPによるDEPTORのユビキチン化はmTORによるDEPTORのリン酸化に依存的であること,すなわち,mTORはDEPTORの分解を促進して自己の活性を増幅させる機構をもつことをあわせて明らかにした.mTORはオートファジーの負の制御タンパク質であると考えられており,この解析においても,SCFβTrCPによるDEPTORの分解を介したmTORの活性制御がオートファジーの調節に密接に関連していた.このことは,SCFβTrCPを介するDEPTOR分解経路が栄養ストレス応答などの細胞の恒常性の維持に重要な役割をはたしていることを示唆している.
免疫抑制剤ラパマイシンの標的タンパク質として同定されたmTOR(mammalian target of rapamycin)は進化的に保存されたセリン-スレオニンキナーゼであり,細胞内外の栄養状態やストレスなどの環境変化を感知しその情報を統合することにより,細胞の成長,増殖,生存のシグナルを下流に伝達する重要な制御装置としての役割を担っている1).mTORは異なる機能をもつ2種類の複合体,mTOR複合体1とmTOR複合体2を形成し,これらの複合体において触媒サブユニットとしてはたらいている(図1).mTOR複合体のおもな機能として,mTOR複合体1はS6キナーゼや4EBP1(eIF4E-binding protein 1)のリン酸化を介したタンパク質合成あるいは細胞成長にかかわるシグナル伝達系を活性化させ,mTOR複合体2はAKTやSGKのリン酸化を介した細胞生存にかかわるシグナル伝達系を活性化させている.さらに,mTOR複合体1はULK1複合体の構成タンパク質であるATG13およびULK1のリン酸化を介してオートファゴソームの形成を抑制しオートファジーを負に調節する2).さまざまな細胞機能の制御をつかさどるmTOR複合体の活性は精密かつ複雑な調節をうけており,mTORのキナーゼ活性を規定する分子機構の解明はmTOR複合体シグナル伝達系の制御異常に起因するさまざまな疾患の病態の解明という点においても重要である.
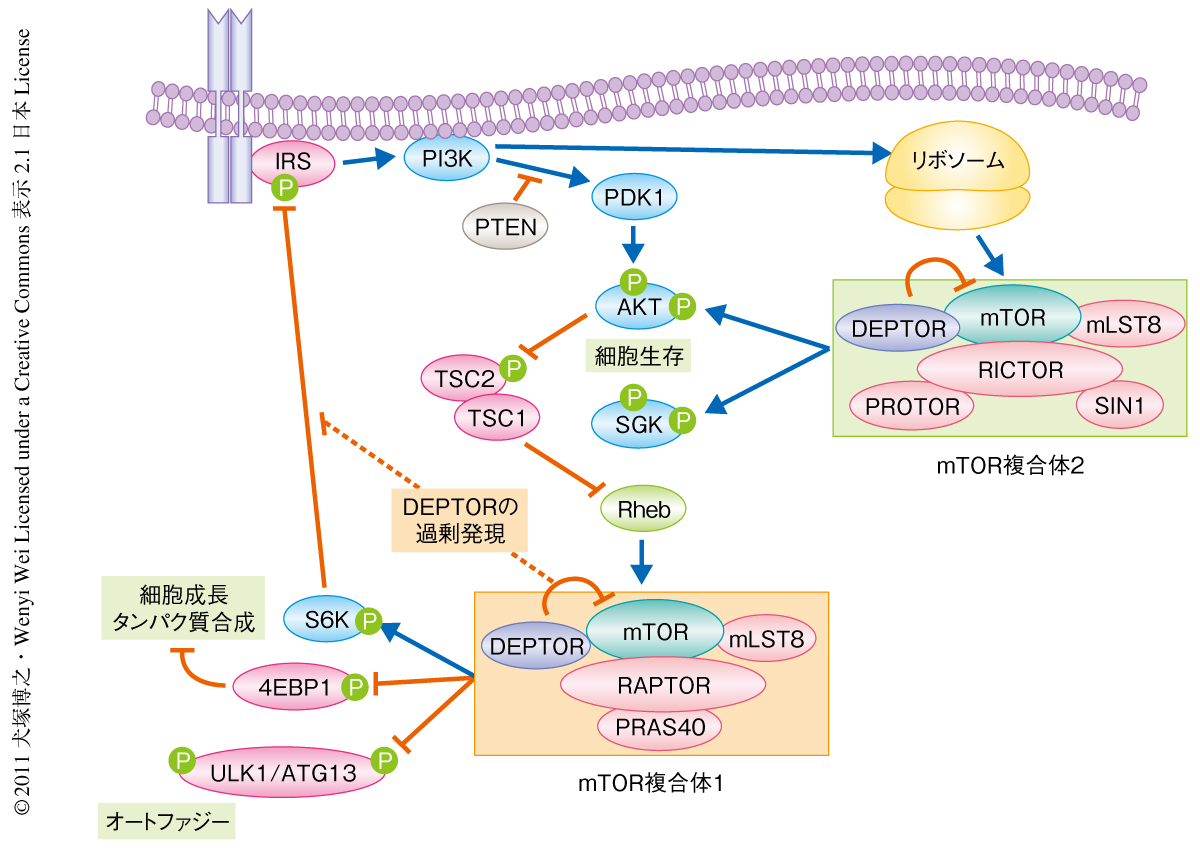
DEPTOR(DEP domain-containing mTOR-interacting protein)はmTORの内在性の阻害タンパク質として,2009年に報告された3).DEPTORはそのC末端側に位置するPDZドメインを介してmTORのFATドメインと結合しmTORの触媒活性を阻害する.DEPTORは低栄養状態の細胞においてmTOR複合体1およびmTOR複合体2の機能を抑制しているが,細胞が増殖因子の刺激を受容するとDEPTORはすみやかにその発現を減少させてmTORの抑制を解除しmTOR複合体シグナル伝達系を活性化させる.増殖因子の刺激により誘導されるDEPTOR発現量の減少はユビキチン-プロテアソーム系に依存的なタンパク質分解経路を介することが示唆されているが3),DEPTOR分解の分子機構はこれまで明らかにされておらず,その役割を担うユビキチンリガーゼもみつかっていなかった.このような背景のもと,DEPTORのユビキチン化による分解の作用機序,および,DEPTORの分解を介したmTORの新たな活性調節機構の解明を目的として解析を行った.
DEPTORがユビキチン-プロテアソーム系を介して分解をうけるとの報告から,まず,プロテオミクス的な手法を用いてDEPTORの分解に関与するタンパク質の同定を試みた.タグを付加したDEPTORを293T細胞において過剰発現してその免疫沈降物を精製し,質量分析法を用いて免疫沈降物に含まれるDEPTORと相互作用したタンパク質を網羅的に解析した.このスクリーニングの過程で,βTrCP2がDEPTORによる免疫沈降物に含まれていることが確認された.βTrCPはSCFβTrCPユビキチンリガーゼ複合体の可変的な基質認識サブユニット(Fボックスタンパク質)であり,βTrCP1とβTrCP2の生化学的に機能の重複する2種類のパラログが存在する4,5).同様の手法により,βTrCP2による免疫沈降によりβTrCP2と結合するタンパク質の同定を試みたところ,既知のβTrCPの基質(βカテニン,CDC25,REST,Claspin,SPAR,Wee1など)とともに,DEPTORを同定することができた.さらに,このβTrCPとDEPTORとの結合はWD40ドメインとよばれるβTrCPのC末端側にある基質認識領域を介していることがわかった.これらの結果から,SCFβTrCPがDEPTORの分解に関与するユビキチンリガーゼであることが強く示唆された.
つぎに,DEPTORの安定性の制御におけるβTrCPの役割を明らかにするため,βTrCPのshRNAによるノックダウンにともなう細胞内のDEPTOR発現量の変化を,mTORシグナル伝達系に及ぼす影響とともに調べた.βTrCPのノックダウンの結果,mTOR複合体を構成するタンパク質のなかで唯一DEPTORの発現のみが有意に上昇し,それにともないmTOR複合体の下流の標的タンパク質であるAKTおよびS6キナーゼのリン酸化の減衰が確認された.さらに,βTrCPのノックダウンによりDEPTORのタンパク質レベルでの半減期の増大が認められた一方,DEPTORのmRNAレベルでの発現量に有意な変化はみられなかったことから,DEPTOR発現量の制御にはタンパク質分解経路を介した翻訳後修飾,すなわち,SCFβTrCPによるDEPTORのユビキチン化が必要であることが示唆された.
βTrCPはリン酸化修飾をうけたβTrCP認識配列に結合することから4,5),各種のキナーゼ阻害剤を用いてβTrCP認識配列を修飾する上流のキナーゼの同定を試みた.キナーゼ阻害剤により処理した細胞に血清刺激をくわえてDEPTORの分解が抑制されるかどうかを検討したところ,対照となる細胞で観察されたDEPTORの分解は,PI3キナーゼの阻害剤LY294002,mTOR複合体の阻害剤PP242,カゼインキナーゼ1の阻害剤D4476による処理により有意に抑制された.さらに,mTOR複合体1およびmTOR複合体2の足場タンパク質として機能するRAPTORおよびRICTORのノックダウン,また,カゼインキナーゼ1αのノックダウンによりDEPTORの細胞内における蓄積が観察された.PP242あるいはD4476により処理した細胞においてDEPTORとβTrCPとの結合が阻害されることから,mTOR複合体1,mTOR複合体2,カゼインキナーゼ1αによるDEPTORのリン酸化がβTrCPとDEPTORとの相互作用を促進し,SCFβTrCPによるDEPTORの分解を誘導するという作用機序が示唆された.
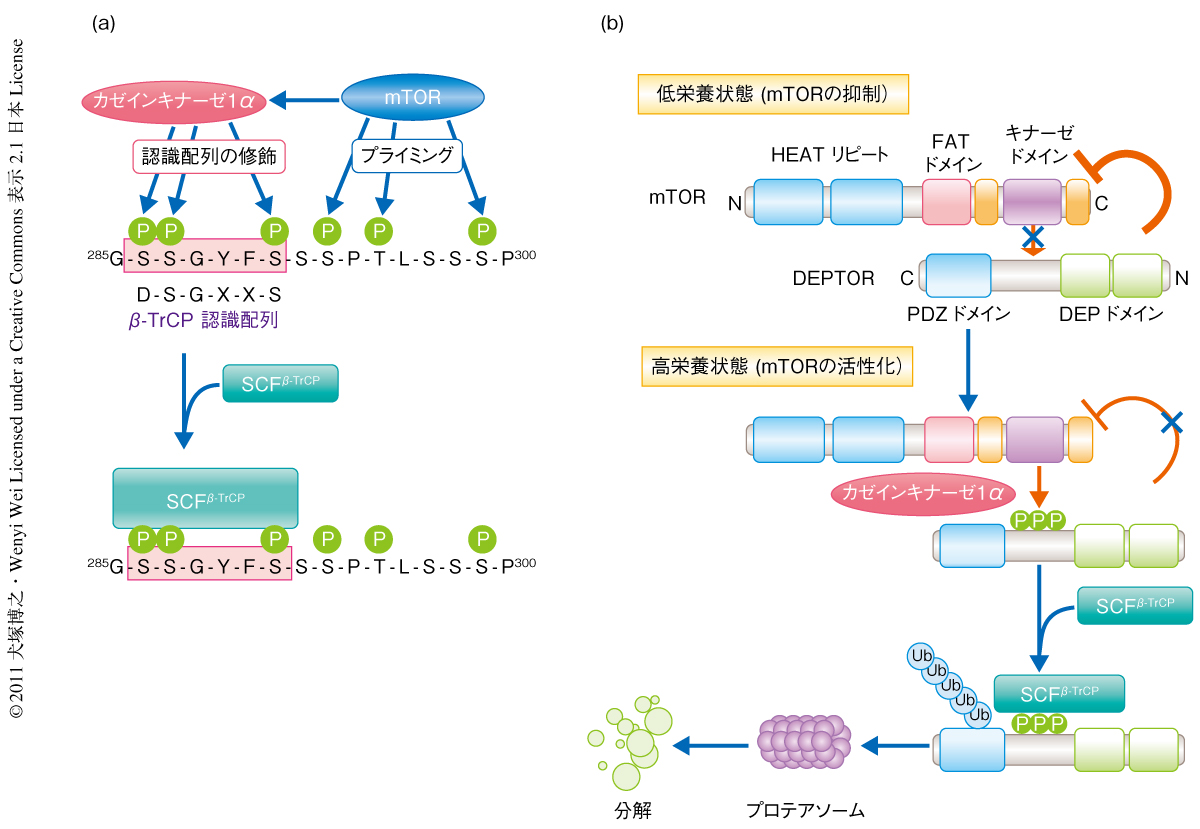
SCFβTrCPによるDEPTOR分解の分子機構の解明のため,DEPTORに存在するβTrCP認識配列のマッピングを行った.βTrCPは,βTrCP認識配列Asp-Ser-Gly-X-X-Serあるいはその類似配列Ser-Ser-Gly/Glu-Ser-Gly/Asp-Asp-Gly-X-X-Ser/Glu/Asp(Xは任意のアミノ酸残基)などを標的とするが5),この配列のSer残基のリン酸化がβTrCPとの結合には必須である.DEPTORのアミノ酸配列の精査の結果,DEPTORにおいてβTrCP認識配列に類似した配列286Ser-Ser-Gly-Tyr-Phe-Ser291を見い出した.質量分析法とin vitroリン酸化アッセイ法による解析から,mTORによりこの配列の近傍にあるSer293,Thr295,Ser299が,カゼインキナーゼ1によりこの配列のSer286,Ser287(ほかの研究グループは,抗リン酸化DEPTOR抗体を用いてさらにSer291を同定した6))が,それぞれリン酸化されることを確認した.これらSer残基とThr残基のうちのいずれかひとつをAla残基に置換することによりβTrCPとDEPTORとの相互作用が阻害されDEPTORの分解は抑制された.以上の結果から,このモチーフが機能的なβTrCP認識配列としてDEPTORの分解にかかわっているものと結論づけた.ちなみに,この配列はすでに報告のあるBimELの分解におけるβTrCP認識配列とまったく同一であった7).
さらに,in vitroでのリン酸化アッセイとユビキチン化アッセイにより,mTORによるこれらのリン酸化がカゼインキナーゼ1によるDEPTORリン酸化の効率を著しく向上させ,それにつづくSCFβTrCPのDEPTORユビキチン化を相乗的に増大させることを確認した.このことは,カゼインキナーゼ1によるβTrCP認識配列のリン酸化修飾のためのプライミング酵素として,mTORが重要な役割をはたしていることを示していた(図2a).ほかの研究グループは,SCFβTrCPによるDEPTORの分解においてS6キナーゼとRSKが協調してβTrCP認識配列を修飾していることを報告しており8),SCFβTrCPが場所や状況に応じてシグナルを使い分けている可能性が考えられたが,この結果は,S6キナーゼの活性を直接に制御するmTORの活性化がDEPTOR分解の引き金をひくという観点において,筆者らの解析結果と一致していた.
mTORは細胞内外のさまざまな環境における栄養やエネルギーのレベルを感知するセンサーとしての役割を担っており,栄養源の枯渇によるmTORの活性低下がオートファジーを誘導することが知られている2).SCFβTrCPに依存的なDEPTOR分解経路の生理的な意義を検討するためオートファジーの活性化を指標に解析を行い,以下の結果を得た.1)βTrCPのノックダウンにともなうDEPTORの細胞内における蓄積により,グルコース存在下の細胞においてもオートファジーが誘導された.2)βTrCPとの結合能を欠失させた安定型DEPTOR変異体を発現誘導させてオートファジーを定量したところ,DEPTORの発現量に相関してオートファジーが誘導された.3)DEPTORのノックダウン細胞および対照となる細胞においてオートファジーの定量を行ったところ,対照細胞においてグルコースの除去により観察されたオートファジーの活性化が,DEPTORのノックダウン細胞において有意に抑制された.以上から,SCFβTrCP-DEPTOR経路を介するmTOR複合体シグナル伝達系の制御が細胞内外の栄養条件の変化にともなうオートファジーの誘導を精密に制御していることが明らかになった.
mTORの内在性の阻害タンパク質DEPTORはSCFβTrCPによるユビキチン化を介したプロテアソームに依存的な分解をうけるほか,このユビキチン化にはカゼインキナーゼ1αとmTORによる協調的なリン酸化が必要であることが明らかになった(図2b).このことは,mTORはSCFβTrCPと連携してDEPTORの分解を誘導することにより,自らの活性を増幅させる正のフィードバックループを形成していることを意味していた.この解析では,DEPTOR分解の生理的な意義を明らかにする目的からオートファジーに焦点をあてたが,mTOR複合体シグナル伝達系は細胞の成長,増殖,生存,老化や代謝などさまざまな機能を制御しており1),SCFβTrCPを介したDEPTOR分解経路がこれら細胞機能にどのように関与しているのか,今後,さらなる解析が必要と思われる.
mTOR複合体シグナル伝達系の亢進がさまざまながんにおいて報告されていることからDEPTORのがん抑制機能が注目されるが,その一方で,多発性骨髄腫においてDEPTORの過剰発現が報告されている3).それらの腫瘍細胞ではDEPTORの過剰発現に起因してPI3キナーゼ-AKT経路が恒常的に活性化されており(図1),細胞生存シグナル伝達系の亢進が観察されている.このことは,mTOR活性とDEPTOR発現との相互のバランスのとれた制御が細胞の正常な増殖および生存に不可欠であることを示唆しており,両者のバランス制御を担うSCFβTrCP-DEPTOR経路の異常がある種のがん化に関連している可能性も考えられる.がん以外の疾患においても,過食による高栄養条件下で恒常的に活性化されるmTORは糖尿病など代謝性の疾患における主要な標的タンパク質と考えられている.さらに近年,mTORやS6キナーゼの機能抑制が延命効果をもたらすことがマウスで報告され1),酵母を基点としたmTORシグナル伝達系の分子レベルでの研究は,哺乳動物の寿命制御にまで広がりをみせている.今回,同定されたSCFβTrCPによるDEPTOR分解経路が新たなmTORの活性調節機構として,がんや糖尿病をはじめとしたさまざまな疾患や個体の老化などにどのようにかかわっているのかも興味深い研究課題であると思われる.
略歴:2000年 東京大学大学院農学生命科学研究科博士課程 修了,同年 千葉県がんセンター 研修生,2001年 香川医科大学医学部 教務職員,2004年 米国Emory大学School of Medicine研究員,2006年 米国Harvard大学Medical School研究員を経て,2010年より同 インストラクター.
研究テーマ:SCFユビキチンリガーゼ複合体の生理機能と発がん.
Wenyi Wei
米国Harvard大学Medical SchoolにてAssistant Professor.
© 2011 犬塚博之・Wenyi Wei Licensed under CC 表示 2.1 日本
(米国Harvard大学Medical School,Beth Israel Deaconess Medical Center,Department of Pathology)
email:犬塚博之
DOI: 10.7875/first.author.2011.174
mTOR drives its own activation via SCFβ-TrCP-dependent degradation of the mTOR inhibitor DEPTOR.
Daming Gao, Hiroyuki Inuzuka, Meng-Kwang Marcus Tan, Hidefumi Fukushima, Jason W. Locasale, Pengda Liu, Lixin Wan, Bo Zhai, Y. Rebecca Chin, Shavali Shaik, Costas A. Lyssiotis, Steven P. Gygi, Alex Toker, Lewis C. Cantley, John M. Asara, J. Wade Harper, Wenyi Wei
Molecular Cell, 44, 290-303 (2011)
要 約
mTORは機能的に異なる2種類の複合体,mTOR複合体1とmTOR複合体2を形成するが,この2つの複合体にはmTOR抑制タンパク質であるDEPTORが含まれている.低栄養状態の細胞ではDEPTORはmTORに直接結合してその活性を抑制しているが,増殖因子の刺激によりDEPTORはすみやかな分解をうけmTOR複合体の活性化を誘導する.DEPTOR分解の分子機構はこれまで不明であったが,今回,筆者らは,ユビキチンリガーゼ複合体SCFβTrCPがDEPTORの分解にかかわっていることを明らかにした.さらに,SCFβTrCPによるDEPTORのユビキチン化はmTORによるDEPTORのリン酸化に依存的であること,すなわち,mTORはDEPTORの分解を促進して自己の活性を増幅させる機構をもつことをあわせて明らかにした.mTORはオートファジーの負の制御タンパク質であると考えられており,この解析においても,SCFβTrCPによるDEPTORの分解を介したmTORの活性制御がオートファジーの調節に密接に関連していた.このことは,SCFβTrCPを介するDEPTOR分解経路が栄養ストレス応答などの細胞の恒常性の維持に重要な役割をはたしていることを示唆している.
はじめに
免疫抑制剤ラパマイシンの標的タンパク質として同定されたmTOR(mammalian target of rapamycin)は進化的に保存されたセリン-スレオニンキナーゼであり,細胞内外の栄養状態やストレスなどの環境変化を感知しその情報を統合することにより,細胞の成長,増殖,生存のシグナルを下流に伝達する重要な制御装置としての役割を担っている1).mTORは異なる機能をもつ2種類の複合体,mTOR複合体1とmTOR複合体2を形成し,これらの複合体において触媒サブユニットとしてはたらいている(図1).mTOR複合体のおもな機能として,mTOR複合体1はS6キナーゼや4EBP1(eIF4E-binding protein 1)のリン酸化を介したタンパク質合成あるいは細胞成長にかかわるシグナル伝達系を活性化させ,mTOR複合体2はAKTやSGKのリン酸化を介した細胞生存にかかわるシグナル伝達系を活性化させている.さらに,mTOR複合体1はULK1複合体の構成タンパク質であるATG13およびULK1のリン酸化を介してオートファゴソームの形成を抑制しオートファジーを負に調節する2).さまざまな細胞機能の制御をつかさどるmTOR複合体の活性は精密かつ複雑な調節をうけており,mTORのキナーゼ活性を規定する分子機構の解明はmTOR複合体シグナル伝達系の制御異常に起因するさまざまな疾患の病態の解明という点においても重要である.
DEPTOR(DEP domain-containing mTOR-interacting protein)はmTORの内在性の阻害タンパク質として,2009年に報告された3).DEPTORはそのC末端側に位置するPDZドメインを介してmTORのFATドメインと結合しmTORの触媒活性を阻害する.DEPTORは低栄養状態の細胞においてmTOR複合体1およびmTOR複合体2の機能を抑制しているが,細胞が増殖因子の刺激を受容するとDEPTORはすみやかにその発現を減少させてmTORの抑制を解除しmTOR複合体シグナル伝達系を活性化させる.増殖因子の刺激により誘導されるDEPTOR発現量の減少はユビキチン-プロテアソーム系に依存的なタンパク質分解経路を介することが示唆されているが3),DEPTOR分解の分子機構はこれまで明らかにされておらず,その役割を担うユビキチンリガーゼもみつかっていなかった.このような背景のもと,DEPTORのユビキチン化による分解の作用機序,および,DEPTORの分解を介したmTORの新たな活性調節機構の解明を目的として解析を行った.
1.DEPTORの分解をつかさどるユビキチンリガーゼの探索
DEPTORがユビキチン-プロテアソーム系を介して分解をうけるとの報告から,まず,プロテオミクス的な手法を用いてDEPTORの分解に関与するタンパク質の同定を試みた.タグを付加したDEPTORを293T細胞において過剰発現してその免疫沈降物を精製し,質量分析法を用いて免疫沈降物に含まれるDEPTORと相互作用したタンパク質を網羅的に解析した.このスクリーニングの過程で,βTrCP2がDEPTORによる免疫沈降物に含まれていることが確認された.βTrCPはSCFβTrCPユビキチンリガーゼ複合体の可変的な基質認識サブユニット(Fボックスタンパク質)であり,βTrCP1とβTrCP2の生化学的に機能の重複する2種類のパラログが存在する4,5).同様の手法により,βTrCP2による免疫沈降によりβTrCP2と結合するタンパク質の同定を試みたところ,既知のβTrCPの基質(βカテニン,CDC25,REST,Claspin,SPAR,Wee1など)とともに,DEPTORを同定することができた.さらに,このβTrCPとDEPTORとの結合はWD40ドメインとよばれるβTrCPのC末端側にある基質認識領域を介していることがわかった.これらの結果から,SCFβTrCPがDEPTORの分解に関与するユビキチンリガーゼであることが強く示唆された.
つぎに,DEPTORの安定性の制御におけるβTrCPの役割を明らかにするため,βTrCPのshRNAによるノックダウンにともなう細胞内のDEPTOR発現量の変化を,mTORシグナル伝達系に及ぼす影響とともに調べた.βTrCPのノックダウンの結果,mTOR複合体を構成するタンパク質のなかで唯一DEPTORの発現のみが有意に上昇し,それにともないmTOR複合体の下流の標的タンパク質であるAKTおよびS6キナーゼのリン酸化の減衰が確認された.さらに,βTrCPのノックダウンによりDEPTORのタンパク質レベルでの半減期の増大が認められた一方,DEPTORのmRNAレベルでの発現量に有意な変化はみられなかったことから,DEPTOR発現量の制御にはタンパク質分解経路を介した翻訳後修飾,すなわち,SCFβTrCPによるDEPTORのユビキチン化が必要であることが示唆された.
2.DEPTORの安定性を制御するキナーゼの同定
βTrCPはリン酸化修飾をうけたβTrCP認識配列に結合することから4,5),各種のキナーゼ阻害剤を用いてβTrCP認識配列を修飾する上流のキナーゼの同定を試みた.キナーゼ阻害剤により処理した細胞に血清刺激をくわえてDEPTORの分解が抑制されるかどうかを検討したところ,対照となる細胞で観察されたDEPTORの分解は,PI3キナーゼの阻害剤LY294002,mTOR複合体の阻害剤PP242,カゼインキナーゼ1の阻害剤D4476による処理により有意に抑制された.さらに,mTOR複合体1およびmTOR複合体2の足場タンパク質として機能するRAPTORおよびRICTORのノックダウン,また,カゼインキナーゼ1αのノックダウンによりDEPTORの細胞内における蓄積が観察された.PP242あるいはD4476により処理した細胞においてDEPTORとβTrCPとの結合が阻害されることから,mTOR複合体1,mTOR複合体2,カゼインキナーゼ1αによるDEPTORのリン酸化がβTrCPとDEPTORとの相互作用を促進し,SCFβTrCPによるDEPTORの分解を誘導するという作用機序が示唆された.
3.リン酸化に依存的なSCFβTrCPによるDEPTOR分解の作用機序
SCFβTrCPによるDEPTOR分解の分子機構の解明のため,DEPTORに存在するβTrCP認識配列のマッピングを行った.βTrCPは,βTrCP認識配列Asp-Ser-Gly-X-X-Serあるいはその類似配列Ser-Ser-Gly/Glu-Ser-Gly/Asp-Asp-Gly-X-X-Ser/Glu/Asp(Xは任意のアミノ酸残基)などを標的とするが5),この配列のSer残基のリン酸化がβTrCPとの結合には必須である.DEPTORのアミノ酸配列の精査の結果,DEPTORにおいてβTrCP認識配列に類似した配列286Ser-Ser-Gly-Tyr-Phe-Ser291を見い出した.質量分析法とin vitroリン酸化アッセイ法による解析から,mTORによりこの配列の近傍にあるSer293,Thr295,Ser299が,カゼインキナーゼ1によりこの配列のSer286,Ser287(ほかの研究グループは,抗リン酸化DEPTOR抗体を用いてさらにSer291を同定した6))が,それぞれリン酸化されることを確認した.これらSer残基とThr残基のうちのいずれかひとつをAla残基に置換することによりβTrCPとDEPTORとの相互作用が阻害されDEPTORの分解は抑制された.以上の結果から,このモチーフが機能的なβTrCP認識配列としてDEPTORの分解にかかわっているものと結論づけた.ちなみに,この配列はすでに報告のあるBimELの分解におけるβTrCP認識配列とまったく同一であった7).
さらに,in vitroでのリン酸化アッセイとユビキチン化アッセイにより,mTORによるこれらのリン酸化がカゼインキナーゼ1によるDEPTORリン酸化の効率を著しく向上させ,それにつづくSCFβTrCPのDEPTORユビキチン化を相乗的に増大させることを確認した.このことは,カゼインキナーゼ1によるβTrCP認識配列のリン酸化修飾のためのプライミング酵素として,mTORが重要な役割をはたしていることを示していた(図2a).ほかの研究グループは,SCFβTrCPによるDEPTORの分解においてS6キナーゼとRSKが協調してβTrCP認識配列を修飾していることを報告しており8),SCFβTrCPが場所や状況に応じてシグナルを使い分けている可能性が考えられたが,この結果は,S6キナーゼの活性を直接に制御するmTORの活性化がDEPTOR分解の引き金をひくという観点において,筆者らの解析結果と一致していた.
4.SCFβTrCPによるDEPTORの分解を介したオートファジーの制御
mTORは細胞内外のさまざまな環境における栄養やエネルギーのレベルを感知するセンサーとしての役割を担っており,栄養源の枯渇によるmTORの活性低下がオートファジーを誘導することが知られている2).SCFβTrCPに依存的なDEPTOR分解経路の生理的な意義を検討するためオートファジーの活性化を指標に解析を行い,以下の結果を得た.1)βTrCPのノックダウンにともなうDEPTORの細胞内における蓄積により,グルコース存在下の細胞においてもオートファジーが誘導された.2)βTrCPとの結合能を欠失させた安定型DEPTOR変異体を発現誘導させてオートファジーを定量したところ,DEPTORの発現量に相関してオートファジーが誘導された.3)DEPTORのノックダウン細胞および対照となる細胞においてオートファジーの定量を行ったところ,対照細胞においてグルコースの除去により観察されたオートファジーの活性化が,DEPTORのノックダウン細胞において有意に抑制された.以上から,SCFβTrCP-DEPTOR経路を介するmTOR複合体シグナル伝達系の制御が細胞内外の栄養条件の変化にともなうオートファジーの誘導を精密に制御していることが明らかになった.
おわりに
mTORの内在性の阻害タンパク質DEPTORはSCFβTrCPによるユビキチン化を介したプロテアソームに依存的な分解をうけるほか,このユビキチン化にはカゼインキナーゼ1αとmTORによる協調的なリン酸化が必要であることが明らかになった(図2b).このことは,mTORはSCFβTrCPと連携してDEPTORの分解を誘導することにより,自らの活性を増幅させる正のフィードバックループを形成していることを意味していた.この解析では,DEPTOR分解の生理的な意義を明らかにする目的からオートファジーに焦点をあてたが,mTOR複合体シグナル伝達系は細胞の成長,増殖,生存,老化や代謝などさまざまな機能を制御しており1),SCFβTrCPを介したDEPTOR分解経路がこれら細胞機能にどのように関与しているのか,今後,さらなる解析が必要と思われる.
mTOR複合体シグナル伝達系の亢進がさまざまながんにおいて報告されていることからDEPTORのがん抑制機能が注目されるが,その一方で,多発性骨髄腫においてDEPTORの過剰発現が報告されている3).それらの腫瘍細胞ではDEPTORの過剰発現に起因してPI3キナーゼ-AKT経路が恒常的に活性化されており(図1),細胞生存シグナル伝達系の亢進が観察されている.このことは,mTOR活性とDEPTOR発現との相互のバランスのとれた制御が細胞の正常な増殖および生存に不可欠であることを示唆しており,両者のバランス制御を担うSCFβTrCP-DEPTOR経路の異常がある種のがん化に関連している可能性も考えられる.がん以外の疾患においても,過食による高栄養条件下で恒常的に活性化されるmTORは糖尿病など代謝性の疾患における主要な標的タンパク質と考えられている.さらに近年,mTORやS6キナーゼの機能抑制が延命効果をもたらすことがマウスで報告され1),酵母を基点としたmTORシグナル伝達系の分子レベルでの研究は,哺乳動物の寿命制御にまで広がりをみせている.今回,同定されたSCFβTrCPによるDEPTOR分解経路が新たなmTORの活性調節機構として,がんや糖尿病をはじめとしたさまざまな疾患や個体の老化などにどのようにかかわっているのかも興味深い研究課題であると思われる.
文 献
- Zoncu, R., Efeyan, A. & Sabatini, D. M.: mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol., 12, 21-35 (2011)[PubMed]
- Levine, B., Mizushima, N. & Virgin, H. W.: Autophagy in immunity and inflammation. Nature, 469, 323-335 (2011)[PubMed]
- Peterson, T. R., Laplante, M., Thoreen, C. C. et al.: DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell, 137, 873-886 (2009)[PubMed]
- Nakayama, K. I. & Nakayama, K.: Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer, 6, 369-381 (2006)[PubMed]
- Frescas, D. & Pagano, M.: Deregulated proteolysis by the F-box proteins SKP2 and β-TrCP: tipping the scales of cancer. Nat. Rev. Cancer, 8, 438-449 (2008)[PubMed]
- Duan, S., Skaar, J. R., Kuchay, S. et al.: mTOR generates an auto-amplification loop by triggering the βTrCP- and CK1α-dependent degradation of DEPTOR. Mol. Cell, 44, 317-324 (2011)[PubMed]
- Dehan, E., Bassermann, F., Guardavaccaro, D. et al.: βTrCP- and Rsk1/2-mediated degradation of BimEL inhibits apoptosis. Mol. Cell, 33, 109-116 (2009)[PubMed]
- Zhao, Y., Xiong, X. & Sun, Y.: DEPTOR, an mTOR inhibitor, is a physiological substrate of SCFβTrCP E3 ubiquitin ligase and regulates survival and autophagy. Mol. Cell, 44, 304-316 (2011)[PubMed]
著者プロフィール
略歴:2000年 東京大学大学院農学生命科学研究科博士課程 修了,同年 千葉県がんセンター 研修生,2001年 香川医科大学医学部 教務職員,2004年 米国Emory大学School of Medicine研究員,2006年 米国Harvard大学Medical School研究員を経て,2010年より同 インストラクター.
研究テーマ:SCFユビキチンリガーゼ複合体の生理機能と発がん.
Wenyi Wei
米国Harvard大学Medical SchoolにてAssistant Professor.
© 2011 犬塚博之・Wenyi Wei Licensed under CC 表示 2.1 日本
