異常タンパク質の選択的オートファジーによる分解はリン酸化p62により制御されている
松本 弦・貫名信行
(理化学研究所脳科学総合研究センター 構造神経病理研究チーム)
email:松本 弦,貫名信行
DOI: 10.7875/first.author.2011.167
Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins.
Gen Matsumoto, Koji Wada, Misako Okuno, Masaru Kurosawa, Nobuyuki Nukina
Molecular Cell, 44, 279-289 (2011)
選択的オートファジーによるポリユビキチン化タンパク質の分解はユビキチン-プロテアソーム系によるタンパク質分解を補完する系であると考えられているが,ポリユビキチン化タンパク質のオートファゴソームへの取り込みを制御する分子機構についてはまったくわかっていない.筆者らは,選択的オートファジーにおける主要なアダプタータンパク質であるp62の403番目のセリン残基がカゼインキナーゼ2によりリン酸化されると,ポリユビキチン化タンパク質のオートファジーによる分解が促進されることを見い出した.このリン酸化によりp62はポリユビキチン鎖との親和性が増強し,ポリユビキチン化タンパク質を“セクエストソーム”のなかに効率よく隔離できるようになった.セクエストソームはそののちオートファゴソームに取り込まれリソソームにより分解された.これらの結果は,オートファジーによる異常タンパク質分解の制御機構の一端を明らかにするとともに,p62による変性タンパク質ストレス応答の分子機構の解明に貢献するものと考えられる.
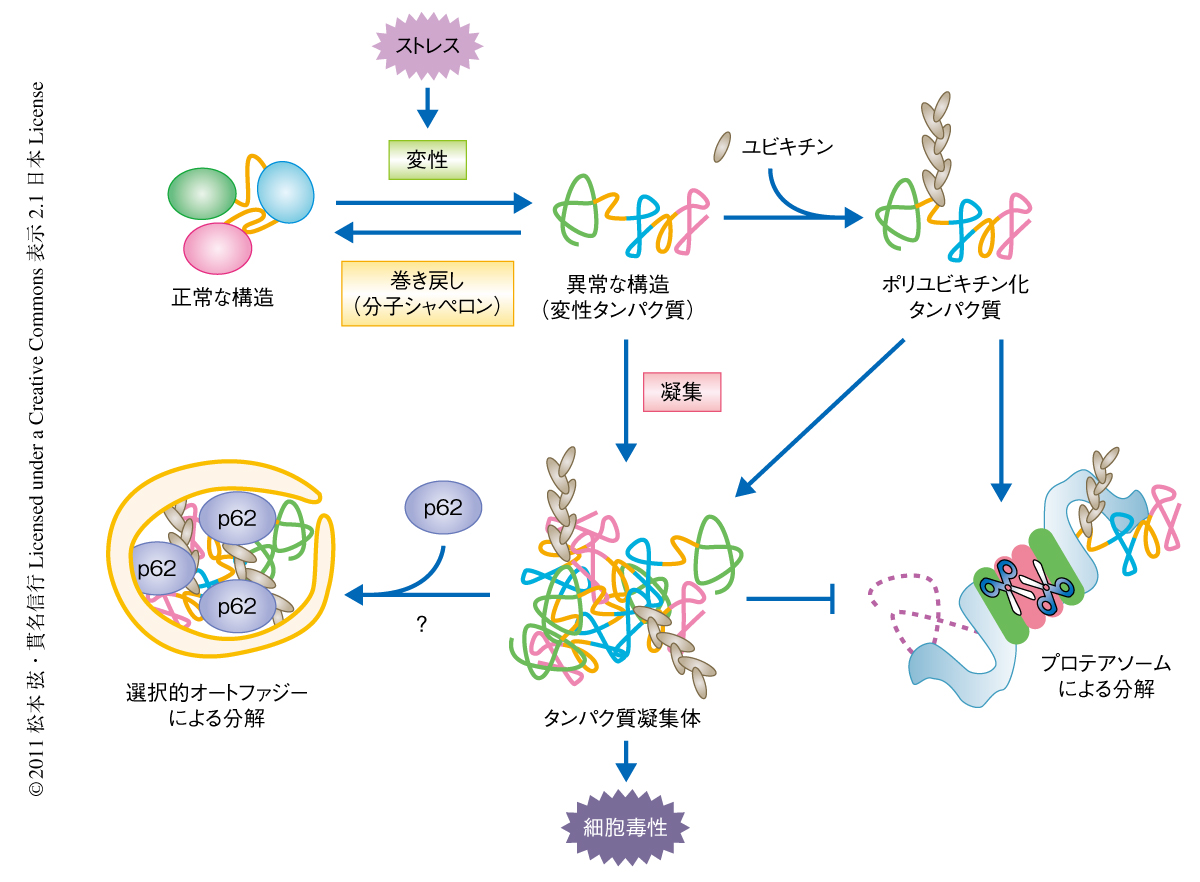
細胞は生きていくためたえずタンパク質をつくり分解するというサイクルをくり返している.細胞には,大別してユビキチン-プロテアソーム系とマクロオートファジー-リソソーム系の2つのタンパク質分解経路が存在する(図1).タンパク質の構造が壊れて機能を失った異常タンパク質のほとんどはユビキチン-プロテアソーム系により分解されるが,プロテアソームには凝集したタンパク質は壊せないという弱点がある1).プロテアソームでは壊せない異常タンパク質の分解はマクロオートファジー-リソソーム系(以下,単にオートファジーという)が担うことになる.オートファジーは,元来,細胞の栄養状態が悪くなり飢餓状態になると細胞質にあるタンパク質やオルガネラを二重の脂質膜で非選択的に取り囲み(オートファゴソーム),リソソームに運んで分解し再利用するというタンパク質分解系である.最近になり,オートファジーは飢餓の誘導時だけでなく低頻度ではあるものの恒常的に起こっていて,ポリユビキチン化タンパク質もオートファジーにより特異的に分解されていることがわかってきた2,3)(選択的オートファジー).オートファゴソームには基質の選択性がないため,基質に特異的なタンパク質分解を行うためには基質とオートファゴソームとをつなぐアダプタータンパク質が必要となる.p62(別名Sequestosome1/SQSTM1/A170)はC末端側にユビキチン結合ドメインであるUBAドメインとオートファゴソームの主要な構成タンパク質のひとつであるLC3との結合ドメインの両方をもつため,ポリユビキチン化タンパク質の選択的オートファジーにおける主要なアダプタータンパク質であると考えられている4).しかしながら,p62のUBAドメインとユビキチンとの親和性は非常に弱く5),ポリユビキチン化タンパク質がどのようにして特異的にオートファジー系へ運ばれるのかという問題は大きな謎であった.今回,筆者らは,p62がリン酸化されることによりポリユビキチン鎖と強く結合できるようになり,ポリユビキチン化タンパク質をオートファジーにより分解するという分子機構を解明した.そして,p62を介した選択的オートファジーがタンパク質の“品質管理”に非常に重要な役割をはたしていることを実証した.
p62が多機能タンパク質であることなどから6,7),p62の機能はリン酸化などの翻訳後修飾により制御されているものと考えた.プロテアソーム阻害の条件においてp62の修飾部位を質量分析により解析したところ,p62が多くの部位においてリン酸化をうけていることを見い出した.これらのリン酸化部位に対する特異的な抗体を作製し,実際にリン酸化が細胞で起こっていることを確認した.オートファゴソームとリソソームとの融合を阻害することで細胞にオートファゴソームを蓄積させると,403番目のセリン残基(S403,以下,アミノ酸残基番号はすべてヒトp62の配列にもとづき表記する)がリン酸化されたp62が顕著に蓄積することを見い出した.また,S403リン酸化p62は脱リン酸化酵素の阻害によっても増加した.これらの結果から,定常状態の細胞においてはS403リン酸化p62とS403非リン酸化p62の両方が平衡状態で共存していると考えられた.
p62は細胞において2つの状態,つまり,細胞質に拡散した状態とp62小体とよばれる構造体を形成した状態として存在するが4),S403リン酸化p62はp62小体に局在することを免疫蛍光法により確認した.S403リン酸化p62を模倣したS403E変異体とS403非リン酸化p62を模倣したS403A変異体を細胞に発現させると,S403E変異p62を発現させた細胞ではp62小体の形成の顕著な亢進がみられた.さらに,S403E変異p62は定常状態においても野生型p62と比べ寿命が短くなっていることもわかった.
p62小体は,p62を含んだオートファゴソーム,それがリソソームと融合したオートファゴリソソーム,膜構造をもたないp62が凝集した“セクエストソーム”の,少なくても3つの異なる構造体の総称である4,8,9).p62のS403リン酸化によりどの構造体の形成が促進するのかを調べるため,Atg5をノックダウンした細胞にS403E変異p62を発現させた.Atg5ノックダウン細胞ではオートファゴソームが形成されないためp62はセクエストソームとして蓄積する.S403E変異p62を発現させた細胞は,野生型p62を発現させた細胞と比べ明らかに多数のセクエストソームを形成した.これらことから,p62はS403リン酸化により効率よくセクエストソームを形成できるようになることが示唆された.
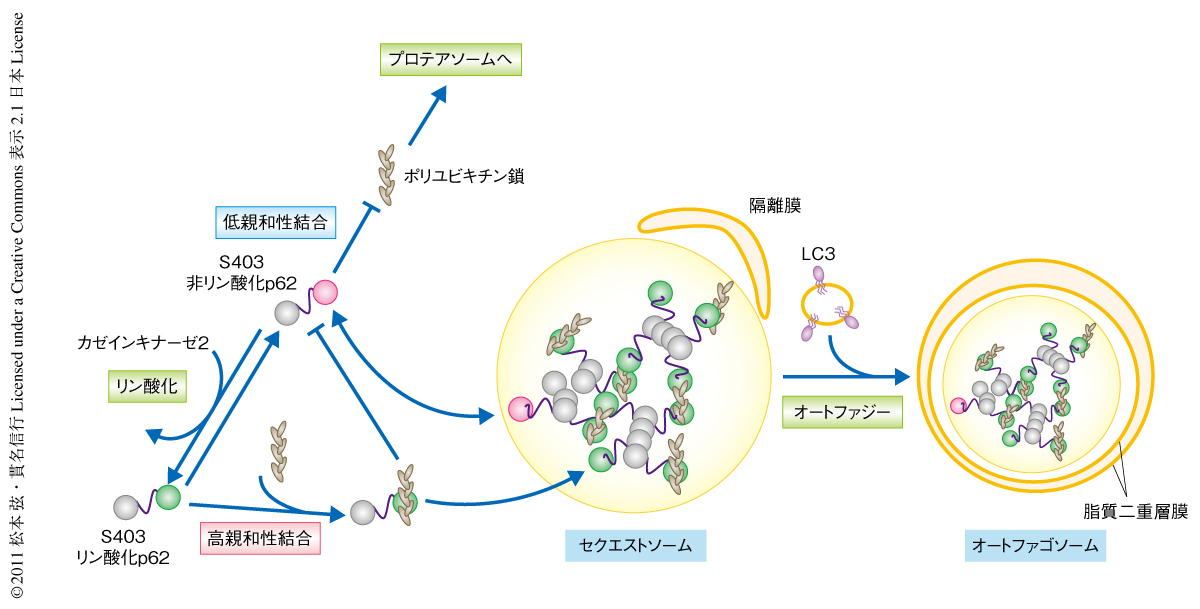
セクエストソームにはポリユビキチン化タンパク質が含まれている8).そこで,S403A変異p62を発現させた細胞とS403E変異p62を発現させた細胞についてセクエストソームとユビキチンの共局在を比較した.その結果,S403A変異p62を発現させた細胞のセクエストソームにユビキチンの蓄積はみられないのに対して,S403E変異p62を発現させた細胞のセクエストソームのほとんどはユビキチンを蓄積させていた.このことは,p62はS403E変異によりユビキチンとの結合力が変化した可能性を示唆した.この可能性を検証するため,精製p62と精製ポリユビキチン鎖との結合をプルダウン解析により生化学的に調べた.その結果,S403E変異p62では野生型p62やS403A変異p62と比べて,ポリユビキチン鎖との親和性の明らかな亢進が認められた.これらの結果から,p62はS403リン酸化によりポリユビキチン鎖と高い親和性をもって結合できるようになり,セクエストソームを形成してそのなかにポリユビキチン化タンパク質を隔離し,セクエストソームごとオートファゴソームに取り込まれるものと考えられた(図2).これまで,セクエストソームは機能不明の細胞内構造体であったが,セクエストソームはS403リン酸化p62によりポリユビキチン化タンパク質をオートファゴソームへ取り込ませるための“運び手”としての役割をはたしていることが明らかになった.
p62はERK2やカゼインキナーゼ2,プロテインキナーゼAによりin vitroでリン酸化されることがすでに報告されている10).これらのプロテインキナーゼがp62をS403リン酸化する可能性があるかどうか調べるためin vitroリン酸化解析を行った.その結果,カゼインキナーゼ2が精製p62をin vitroでS403リン酸化できることを見い出した.細胞をカゼインキナーゼ2の阻害剤で処理するとp62のS403リン酸化は抑制され,カゼインキナーゼ2の過剰発現によりp62のS403リン酸化は亢進しその分解も促進された.カゼインキナーゼ2は恒常的にキナーゼ活性をもっているため,定常状態においても細胞にS403リン酸化p62が存在するという結果とも矛盾しない.これらのことから,カゼインキナーゼ2が細胞においてp62をS403リン酸化するプロテインキナーゼのひとつであることが示された.
多くの神経変性疾患ではポリユビキチン化された異常タンパク質が細胞に蓄積している.したがって,p62のS403リン酸化を促進することで選択的オートファジーを亢進することができれば,異常タンパク質を効率よく分解し疾患の治療につながることが期待できる.その可能性を検証するため,ハンチントン病の病因遺伝子産物であるハンチンチンの一部を発現したモデル細胞において,カゼインキナーゼ2の過剰発現によりp62のS403リン酸化を促進させた場合の異常ハンチンチンの蓄積の変化を調べた.その結果,p62のS403リン酸化の促進により異常ハンチンチンの凝集体の形成は顕著に抑制された.カゼインキナーゼ2の過剰発現による凝集体の形成の抑制効果はp62をRNAi法によりノックダウンすることで解消されたことから,p62のS403リン酸化による効果であることも確認された.これらの結果は,p62のS403リン酸化の促進により細胞毒性をもつ異常タンパク質を選択的に除去できる可能性を示した.
異常タンパク質が細胞に蓄積するという事態は細胞がストレス応答をひき起こすトリガーとなる.神経細胞は生涯にわたり分裂しないため,細胞毒性をもつ変性タンパク質の蓄積はとくに深刻な問題となる.実際,アルツハイマー病,パーキンソン病,筋萎縮性側索硬化症,ハンチントン病など,多くの神経変性疾患に共通した特徴として,神経細胞に凝集した異常タンパク質の蓄積が観察されている.したがって,神経変性疾患の病因を解明するためには,選択的オートファジーによる異常タンパク質の分解の詳細な分子機構を理解する必要がある.
今回,筆者らが明らかにした選択的オートファジーの制御機構により,つぎの3つの大きな問題が解決した.1つ目は,p62とユビキチンとの結合力が弱いことからp62が本当に選択的オートファジーのアダプタータンパク質として機能しているのかという問題である.p62はS403リン酸化されることによりユビキチンとの結合力が強くなる.この発見により,p62がポリユビキチン化タンパク質とオートファゴソームとをつなぐアダプタータンパク質として機能していることが証明された.2つ目は,ポリユビキチン化タンパク質がどのようにしてプロテアソームによる分解と競合せず選択的にオートファゴソームへ取り込まれるのかという問題である.定常状態において,細胞にはS403リン酸化p62(高親和性ユビキチン結合型)とS403非リン酸化p62(低親和性ユビキチン結合型)がつねに存在する.S403リン酸化p62はポリユビキチン化タンパク質と結合していない場合には脱リン酸化反応によりS403非リン酸化p62へともどるため,高親和性ユビキチン結合型であるS403リン酸化p62は低い濃度で維持されていると思われる.そのため,ほとんどのp62はポリユビキチン化タンパク質と結合せずプロテアソームへのターゲティングの過程には影響しないと考えられる.そして3つ目が,選択的オートファジーの制御が可能かどうかという問題である.ポリユビキチン化タンパク質のオートファゴソームへの輸送はp62のS403リン酸化の状態により制御されていることがわかったので,p62のS403リン酸化を特異的に促進する薬剤を開発することができれば,異常タンパク質の細胞内への蓄積を特徴とするアルツハイマー病,パーキンソン病,ポリグルタミン病,筋萎縮性側索硬化症など,さまざまな神経変性疾患の治療への応用も期待できる.
略歴:2000年 京都大学大学院理学研究科博士課程 修了,同年 米国Northwestern大学Human Frontier Science Program Long-term Fellowを経て,2005年より理化学研究所脳科学総合研究センター 研究員.
研究テーマ:ストレス応答とタンパク質分解の分子機構.
関心事:子どもたちの成長.
貫名 信行(Nobuyuki Nukina)
理化学研究所脳科学総合研究センター チームリーダー.
研究室URL:http://www.brain.riken.jp/labs/cagrds/indexj.html
© 2011 松本 弦・貫名信行 Licensed under CC 表示 2.1 日本
(理化学研究所脳科学総合研究センター 構造神経病理研究チーム)
email:松本 弦,貫名信行
DOI: 10.7875/first.author.2011.167
Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins.
Gen Matsumoto, Koji Wada, Misako Okuno, Masaru Kurosawa, Nobuyuki Nukina
Molecular Cell, 44, 279-289 (2011)
この論文に出現する遺伝子・タンパク質のUniprot ID
p62(Q13501), SQSTM1(Q13501), ポリユビキチン, ユビキチン, プロテアソーム, カゼインキナーゼ2, Sequestosome1(Q13501), A170(Q13501), LC3, Atg5(Q9H1Y0), ERK2(P63085), プロテインキナーゼA, プロテインキナーゼ, ハンチンチン(P42858)
要 約
選択的オートファジーによるポリユビキチン化タンパク質の分解はユビキチン-プロテアソーム系によるタンパク質分解を補完する系であると考えられているが,ポリユビキチン化タンパク質のオートファゴソームへの取り込みを制御する分子機構についてはまったくわかっていない.筆者らは,選択的オートファジーにおける主要なアダプタータンパク質であるp62の403番目のセリン残基がカゼインキナーゼ2によりリン酸化されると,ポリユビキチン化タンパク質のオートファジーによる分解が促進されることを見い出した.このリン酸化によりp62はポリユビキチン鎖との親和性が増強し,ポリユビキチン化タンパク質を“セクエストソーム”のなかに効率よく隔離できるようになった.セクエストソームはそののちオートファゴソームに取り込まれリソソームにより分解された.これらの結果は,オートファジーによる異常タンパク質分解の制御機構の一端を明らかにするとともに,p62による変性タンパク質ストレス応答の分子機構の解明に貢献するものと考えられる.
はじめに
細胞は生きていくためたえずタンパク質をつくり分解するというサイクルをくり返している.細胞には,大別してユビキチン-プロテアソーム系とマクロオートファジー-リソソーム系の2つのタンパク質分解経路が存在する(図1).タンパク質の構造が壊れて機能を失った異常タンパク質のほとんどはユビキチン-プロテアソーム系により分解されるが,プロテアソームには凝集したタンパク質は壊せないという弱点がある1).プロテアソームでは壊せない異常タンパク質の分解はマクロオートファジー-リソソーム系(以下,単にオートファジーという)が担うことになる.オートファジーは,元来,細胞の栄養状態が悪くなり飢餓状態になると細胞質にあるタンパク質やオルガネラを二重の脂質膜で非選択的に取り囲み(オートファゴソーム),リソソームに運んで分解し再利用するというタンパク質分解系である.最近になり,オートファジーは飢餓の誘導時だけでなく低頻度ではあるものの恒常的に起こっていて,ポリユビキチン化タンパク質もオートファジーにより特異的に分解されていることがわかってきた2,3)(選択的オートファジー).オートファゴソームには基質の選択性がないため,基質に特異的なタンパク質分解を行うためには基質とオートファゴソームとをつなぐアダプタータンパク質が必要となる.p62(別名Sequestosome1/SQSTM1/A170)はC末端側にユビキチン結合ドメインであるUBAドメインとオートファゴソームの主要な構成タンパク質のひとつであるLC3との結合ドメインの両方をもつため,ポリユビキチン化タンパク質の選択的オートファジーにおける主要なアダプタータンパク質であると考えられている4).しかしながら,p62のUBAドメインとユビキチンとの親和性は非常に弱く5),ポリユビキチン化タンパク質がどのようにして特異的にオートファジー系へ運ばれるのかという問題は大きな謎であった.今回,筆者らは,p62がリン酸化されることによりポリユビキチン鎖と強く結合できるようになり,ポリユビキチン化タンパク質をオートファジーにより分解するという分子機構を解明した.そして,p62を介した選択的オートファジーがタンパク質の“品質管理”に非常に重要な役割をはたしていることを実証した.
1.p62の403番目のセリン残基のリン酸化は選択的オートファジーを促進する
p62が多機能タンパク質であることなどから6,7),p62の機能はリン酸化などの翻訳後修飾により制御されているものと考えた.プロテアソーム阻害の条件においてp62の修飾部位を質量分析により解析したところ,p62が多くの部位においてリン酸化をうけていることを見い出した.これらのリン酸化部位に対する特異的な抗体を作製し,実際にリン酸化が細胞で起こっていることを確認した.オートファゴソームとリソソームとの融合を阻害することで細胞にオートファゴソームを蓄積させると,403番目のセリン残基(S403,以下,アミノ酸残基番号はすべてヒトp62の配列にもとづき表記する)がリン酸化されたp62が顕著に蓄積することを見い出した.また,S403リン酸化p62は脱リン酸化酵素の阻害によっても増加した.これらの結果から,定常状態の細胞においてはS403リン酸化p62とS403非リン酸化p62の両方が平衡状態で共存していると考えられた.
2.S403リン酸化p62はセクエストソームにポリユビキチン化タンパク質を閉じ込めオートファゴソームへ受け渡す
p62は細胞において2つの状態,つまり,細胞質に拡散した状態とp62小体とよばれる構造体を形成した状態として存在するが4),S403リン酸化p62はp62小体に局在することを免疫蛍光法により確認した.S403リン酸化p62を模倣したS403E変異体とS403非リン酸化p62を模倣したS403A変異体を細胞に発現させると,S403E変異p62を発現させた細胞ではp62小体の形成の顕著な亢進がみられた.さらに,S403E変異p62は定常状態においても野生型p62と比べ寿命が短くなっていることもわかった.
p62小体は,p62を含んだオートファゴソーム,それがリソソームと融合したオートファゴリソソーム,膜構造をもたないp62が凝集した“セクエストソーム”の,少なくても3つの異なる構造体の総称である4,8,9).p62のS403リン酸化によりどの構造体の形成が促進するのかを調べるため,Atg5をノックダウンした細胞にS403E変異p62を発現させた.Atg5ノックダウン細胞ではオートファゴソームが形成されないためp62はセクエストソームとして蓄積する.S403E変異p62を発現させた細胞は,野生型p62を発現させた細胞と比べ明らかに多数のセクエストソームを形成した.これらことから,p62はS403リン酸化により効率よくセクエストソームを形成できるようになることが示唆された.
3.p62はS403リン酸化によりポリユビキチン鎖との親和性が増大する
セクエストソームにはポリユビキチン化タンパク質が含まれている8).そこで,S403A変異p62を発現させた細胞とS403E変異p62を発現させた細胞についてセクエストソームとユビキチンの共局在を比較した.その結果,S403A変異p62を発現させた細胞のセクエストソームにユビキチンの蓄積はみられないのに対して,S403E変異p62を発現させた細胞のセクエストソームのほとんどはユビキチンを蓄積させていた.このことは,p62はS403E変異によりユビキチンとの結合力が変化した可能性を示唆した.この可能性を検証するため,精製p62と精製ポリユビキチン鎖との結合をプルダウン解析により生化学的に調べた.その結果,S403E変異p62では野生型p62やS403A変異p62と比べて,ポリユビキチン鎖との親和性の明らかな亢進が認められた.これらの結果から,p62はS403リン酸化によりポリユビキチン鎖と高い親和性をもって結合できるようになり,セクエストソームを形成してそのなかにポリユビキチン化タンパク質を隔離し,セクエストソームごとオートファゴソームに取り込まれるものと考えられた(図2).これまで,セクエストソームは機能不明の細胞内構造体であったが,セクエストソームはS403リン酸化p62によりポリユビキチン化タンパク質をオートファゴソームへ取り込ませるための“運び手”としての役割をはたしていることが明らかになった.
4.カゼインキナーゼ2はp62をS403リン酸化する
p62はERK2やカゼインキナーゼ2,プロテインキナーゼAによりin vitroでリン酸化されることがすでに報告されている10).これらのプロテインキナーゼがp62をS403リン酸化する可能性があるかどうか調べるためin vitroリン酸化解析を行った.その結果,カゼインキナーゼ2が精製p62をin vitroでS403リン酸化できることを見い出した.細胞をカゼインキナーゼ2の阻害剤で処理するとp62のS403リン酸化は抑制され,カゼインキナーゼ2の過剰発現によりp62のS403リン酸化は亢進しその分解も促進された.カゼインキナーゼ2は恒常的にキナーゼ活性をもっているため,定常状態においても細胞にS403リン酸化p62が存在するという結果とも矛盾しない.これらのことから,カゼインキナーゼ2が細胞においてp62をS403リン酸化するプロテインキナーゼのひとつであることが示された.
5.p62のS403リン酸化を促進することにより異常タンパク質の凝集体の形成を抑制できる
多くの神経変性疾患ではポリユビキチン化された異常タンパク質が細胞に蓄積している.したがって,p62のS403リン酸化を促進することで選択的オートファジーを亢進することができれば,異常タンパク質を効率よく分解し疾患の治療につながることが期待できる.その可能性を検証するため,ハンチントン病の病因遺伝子産物であるハンチンチンの一部を発現したモデル細胞において,カゼインキナーゼ2の過剰発現によりp62のS403リン酸化を促進させた場合の異常ハンチンチンの蓄積の変化を調べた.その結果,p62のS403リン酸化の促進により異常ハンチンチンの凝集体の形成は顕著に抑制された.カゼインキナーゼ2の過剰発現による凝集体の形成の抑制効果はp62をRNAi法によりノックダウンすることで解消されたことから,p62のS403リン酸化による効果であることも確認された.これらの結果は,p62のS403リン酸化の促進により細胞毒性をもつ異常タンパク質を選択的に除去できる可能性を示した.
おわりに
異常タンパク質が細胞に蓄積するという事態は細胞がストレス応答をひき起こすトリガーとなる.神経細胞は生涯にわたり分裂しないため,細胞毒性をもつ変性タンパク質の蓄積はとくに深刻な問題となる.実際,アルツハイマー病,パーキンソン病,筋萎縮性側索硬化症,ハンチントン病など,多くの神経変性疾患に共通した特徴として,神経細胞に凝集した異常タンパク質の蓄積が観察されている.したがって,神経変性疾患の病因を解明するためには,選択的オートファジーによる異常タンパク質の分解の詳細な分子機構を理解する必要がある.
今回,筆者らが明らかにした選択的オートファジーの制御機構により,つぎの3つの大きな問題が解決した.1つ目は,p62とユビキチンとの結合力が弱いことからp62が本当に選択的オートファジーのアダプタータンパク質として機能しているのかという問題である.p62はS403リン酸化されることによりユビキチンとの結合力が強くなる.この発見により,p62がポリユビキチン化タンパク質とオートファゴソームとをつなぐアダプタータンパク質として機能していることが証明された.2つ目は,ポリユビキチン化タンパク質がどのようにしてプロテアソームによる分解と競合せず選択的にオートファゴソームへ取り込まれるのかという問題である.定常状態において,細胞にはS403リン酸化p62(高親和性ユビキチン結合型)とS403非リン酸化p62(低親和性ユビキチン結合型)がつねに存在する.S403リン酸化p62はポリユビキチン化タンパク質と結合していない場合には脱リン酸化反応によりS403非リン酸化p62へともどるため,高親和性ユビキチン結合型であるS403リン酸化p62は低い濃度で維持されていると思われる.そのため,ほとんどのp62はポリユビキチン化タンパク質と結合せずプロテアソームへのターゲティングの過程には影響しないと考えられる.そして3つ目が,選択的オートファジーの制御が可能かどうかという問題である.ポリユビキチン化タンパク質のオートファゴソームへの輸送はp62のS403リン酸化の状態により制御されていることがわかったので,p62のS403リン酸化を特異的に促進する薬剤を開発することができれば,異常タンパク質の細胞内への蓄積を特徴とするアルツハイマー病,パーキンソン病,ポリグルタミン病,筋萎縮性側索硬化症など,さまざまな神経変性疾患の治療への応用も期待できる.
文 献
- Venkatraman, P., Wetzel, R., Tanaka, M. et al.: Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol. Cell, 14, 95-104 (2004)[PubMed]
- Hara, T., Nakamura, K., Matsui, M. et al.: Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature, 441, 885-889 (2006)[PubMed]
- Komatsu, M., Waguri, S., Chiba, T. et al.: Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature, 441, 880-884 (2006)[PubMed]
- Bjorkoy, G., Lamark, T., Brech, A. et al.: p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol., 171, 603-614 (2005)[PubMed]
- Long, J., Gallagher, T. R. A., Cavey, J. R. et al.: Ubiquitin recognition by the ubiquitin-associated domain of p62 involves a novel conformational switch. J. Biol. Chem., 283, 5427-5440 (2008)[PubMed]
- Seibenhener, M. L., Geetha, T. & Wooten, M. W.: Sequestosome 1/p62: more than just a scaffold. FEBS Lett., 581, 175-179 (2007)[PubMed]
- Moscat, J., Diaz-Meco, M. T. & Wooten, M. W.: Signal integration and diversification through the p62 scaffold protein. Trends Biochem. Sci., 32, 95-100 (2007)[PubMed]
- Shin, J.: P62 and the sequestosome, a novel mechanism for protein metabolism. Arch. Pharm Res., 21, 629-633 (1998)[PubMed]
- Pankiv, S., Clausen, T. H., Lamark, T.: p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem., 282, 24131-24145 (2007)[PubMed]
- Yanagawa, T., Yuki, K., Yoshida, H. et al.: Phosphorylation of A170 stress protein by casein kinase II-like activity in macrophages. Biochem. Biophys. Res. Commun., 241, 157-163 (1997)[PubMed]
著者プロフィール
略歴:2000年 京都大学大学院理学研究科博士課程 修了,同年 米国Northwestern大学Human Frontier Science Program Long-term Fellowを経て,2005年より理化学研究所脳科学総合研究センター 研究員.
研究テーマ:ストレス応答とタンパク質分解の分子機構.
関心事:子どもたちの成長.
貫名 信行(Nobuyuki Nukina)
理化学研究所脳科学総合研究センター チームリーダー.
研究室URL:http://www.brain.riken.jp/labs/cagrds/indexj.html
© 2011 松本 弦・貫名信行 Licensed under CC 表示 2.1 日本
