FBXL5-IRP2系は生体における鉄代謝の制御に不可欠である
諸石寿朗・中山敬一
(九州大学生体防御医学研究所 分子医科学分野)
email:諸石寿朗
DOI: 10.7875/first.author.2011.144
The FBXL5-IRP2 axis is integral to control of iron metabolism in vivo.
Toshiro Moroishi, Masaaki Nishiyama, Yukiko Takeda, Kazuhiro Iwai, Keiichi I. Nakayama
Cell Metabolism, 14, 339-351 (2011)
FBXL5はSCF複合体型ユビキチンリガーゼの基質認識サブユニットであり,細胞において鉄の量が増加した場合に鉄代謝制御タンパク質IRP2の分解を促進すると考えられているが,生体における鉄代謝の制御にFBXL5のはたす役割はほとんど理解されていない.筆者らは,マウスの全身においてFBXL5を欠損させるとIRP2の過剰な活性化が起こり,鉄の蓄積により胎生早期に大規模なアポトーシスを起こして致死となることを発見した.この胎生期における致死性はIRP2との二重欠損により回避されたため,FBXL5を欠損したマウスのおもな死因はIRP2の過剰な活性化に起因するものと考えられた.また,肝臓に特異的にFBXL5を欠損させると生まれたマウスは鉄過剰症による脂肪性肝炎を発症し,さらに,高鉄含有食をあたえると1日で急性肝不全を発症し死亡した.以上の結果から,細胞への鉄の適切な供給を確保するというFBXL5の主要な役割が明らかになった.
鉄は電子の授受を容易に行いうることから種々の酵素の活性中心としてはたらいており,地球上のほぼすべての生物にとりその生存に必須な元素である.一方で,過剰に二価鉄(Fe2+)が存在すると,その高い反応性ゆえにフリーラジカルの産生を促進し細胞に対する傷害性をもたらす.つまり,鉄は不足しても過剰でも生体に悪影響を及ぼすため,生体においては鉄の量がつねに適切な量になるよう厳密に調節される必要がある.
細胞における鉄恒常性は,鉄の取り込み,利用,貯蔵,および,排出の協調的な制御により維持されている1)(図1).鉄はFe2+のかたちで細胞に入り,タンパク質への直接的な取り込みや,鉄の利用の主要な器官であるミトコンドリアへの供給に必要な細胞質鉄プールとして蓄積される.利用されない細胞質鉄プールのFe2+は,鉄を介した細胞の損傷を防ぐためフェリチンと結合して酸化還元反応を起こさない三価鉄(Fe3+)として貯蔵されるか,あるいは,フェロポーチンにより細胞外に排出される.
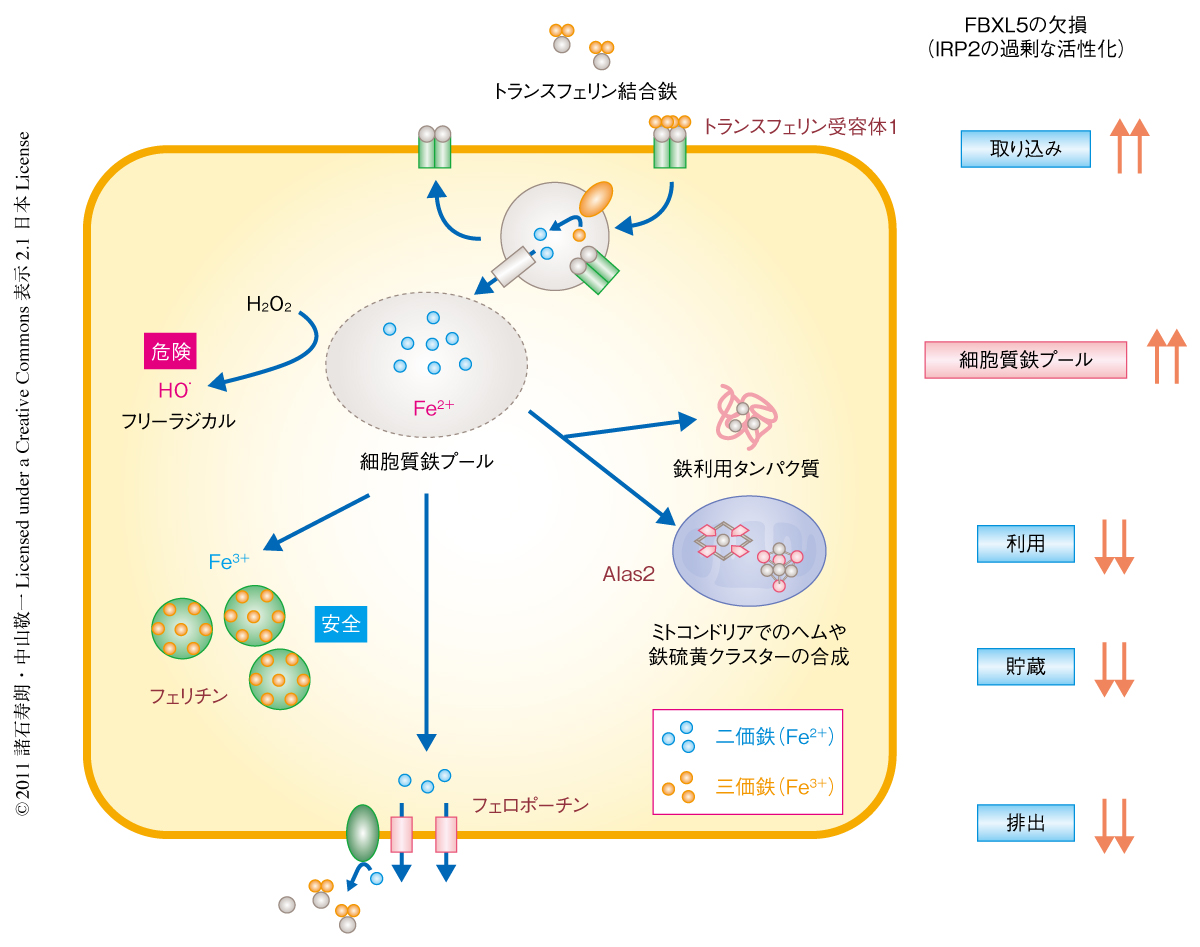
これらの鉄代謝関連タンパク質の発現量はIRP2(iron-regulatory protein 2)により協調的に制御されている2).IRP2はmRNA結合タンパク質であり,鉄の欠乏条件においてフェリチンやフェロポーチン,または,Alas2(aminolevulinic acid synthase 2)のmRNAの5’側非翻訳領域に結合して翻訳の開始を阻害するとともに,トランスフェリン受容体1のmRNAの3’側非翻訳領域に結合してその分解を防ぐ.これらの相互作用により,IRP2は鉄の欠乏条件においてフェリチン(鉄の貯蔵を行う)やフェロポーチン(鉄の排出を行う),Alas2(鉄の利用を行う)の発現量を低下させ,逆に,トランスフェリン受容体1(鉄の取り込みを行う)の発現量を増加させることで,結果として細胞質鉄プールのサイズを大きくする.一方,鉄の過剰条件ではIRP2はSCF複合体型ユビキチンリガーゼSCFFBXL5により分解される3,4).SCFFBXL5の基質認識サブユニットFBXL5(F-box and leucine-rich repeat protein 5)は鉄の欠乏時には不安定であるが,過剰時には自らのもつ鉄結合ドメインへの鉄の配位により安定化しIRP2を分解できるようになる.このように,FBXL5は鉄の過剰条件でのIRP2の分解において不可欠な役割をもつにもかかわらず,生体における鉄恒常性の維持にFBXL5のはたす役割はまったく明らかになっていなかった.今回,筆者らは,FBXL5-IRP2系が生体における鉄恒常性の維持の中心的な制御機構であることを明らかにした.
鉄代謝の制御におけるFBXL5の機能を解析するため,全身においてFbxl5遺伝子を欠損させたFBXL5ノックアウトマウスを作製した.FBXL5ノックアウトマウスの胚は胎生7.5日までは正常に発育したが,胎生8.5日以降には発育遅延と大規模な出血を示し死亡した.組織病理学的な解析を行うと,FBXL5ノックアウトマウスの胚では早期胎盤領域においてFe2+の過剰な蓄積とそれにともなう酸化傷害を認め,凝縮核をともなうアポトーシス細胞が観察された.つぎに,胎盤の機能とは無関係にFBXL5ノックアウトマウスの胚の成長能力を直接に評価するため,胚盤胞をin vitroで培養しその成長を調べた.通常の鉄条件において培養すると,野生型マウスおよびFBXL5ノックアウトマウスの胚盤胞はともに正常に発育した.しかしながら,鉄の過剰条件において培養すると,野生型マウスの胚盤胞は正常に発育したのに対し,FBXL5ノックアウトマウスの胚盤胞の発育は障害された.そして,この発育障害は抗酸化作用のあるN-アセチル-L-システインの投与により回避された.これらの結果から,FBXL5ノックアウトマウスでは鉄過剰ストレスに対する防御機構が破綻しており,Fe2+の過剰な蓄積にともなう酸化ストレスにより傷害をうけていることがわかった.
FBXL5ノックアウトマウスの胚において鉄の過剰な蓄積が観察されたため,IRP2の異常な活性化が鉄恒常性の破綻の原因であるかもしれないという仮説をたてた.実際,FBXL5ノックアウトマウスの胚ではIRP2の下流の標的であるトランスフェリン受容体1のmRNA量が著しく増加しており,IRP2の活性の増加していることが示唆された.FBXL5ノックアウトマウスは胎生早期に致死となるため,鉄恒常性の維持におけるFBXL5の役割を詳細に解析することはむずかしかった.そこで,Fbxl5遺伝子を組織特異的に欠損するようなコンディショナルノックアウトマウスを作製した.このノックアウトマウスから胎仔線維芽細胞を調製しFbxl5遺伝子を欠損させたところ,IRP2の鉄濃度に依存的な分解が障害され,通常はIRP2が分解されるべき鉄過剰の条件においてもIRP2が蓄積していた.この結果と一致して,IRP2の標的となるmRNAのコードするタンパク質の発現量もまたその制御に異常をきたしていた.すなわち,トランスフェリン受容体1の発現量は増加しており,一方,フェリチンの発現量は減少していた.これらの結果は,FBXL5を欠損したマウス胎仔または胎仔線維芽細胞では,細胞における鉄の状態とは無関係にIRP2が蓄積した結果,IRP2の標的の制御に異常をきたしていることを示唆していた(図1).つまり,FBXL5ノックアウトマウスではIRP2が過剰に活性化し,細胞質鉄プールの増大からFe2+の過剰な蓄積とそれにともなう酸化ストレスをきたし,胎生期に死亡することが示唆された.
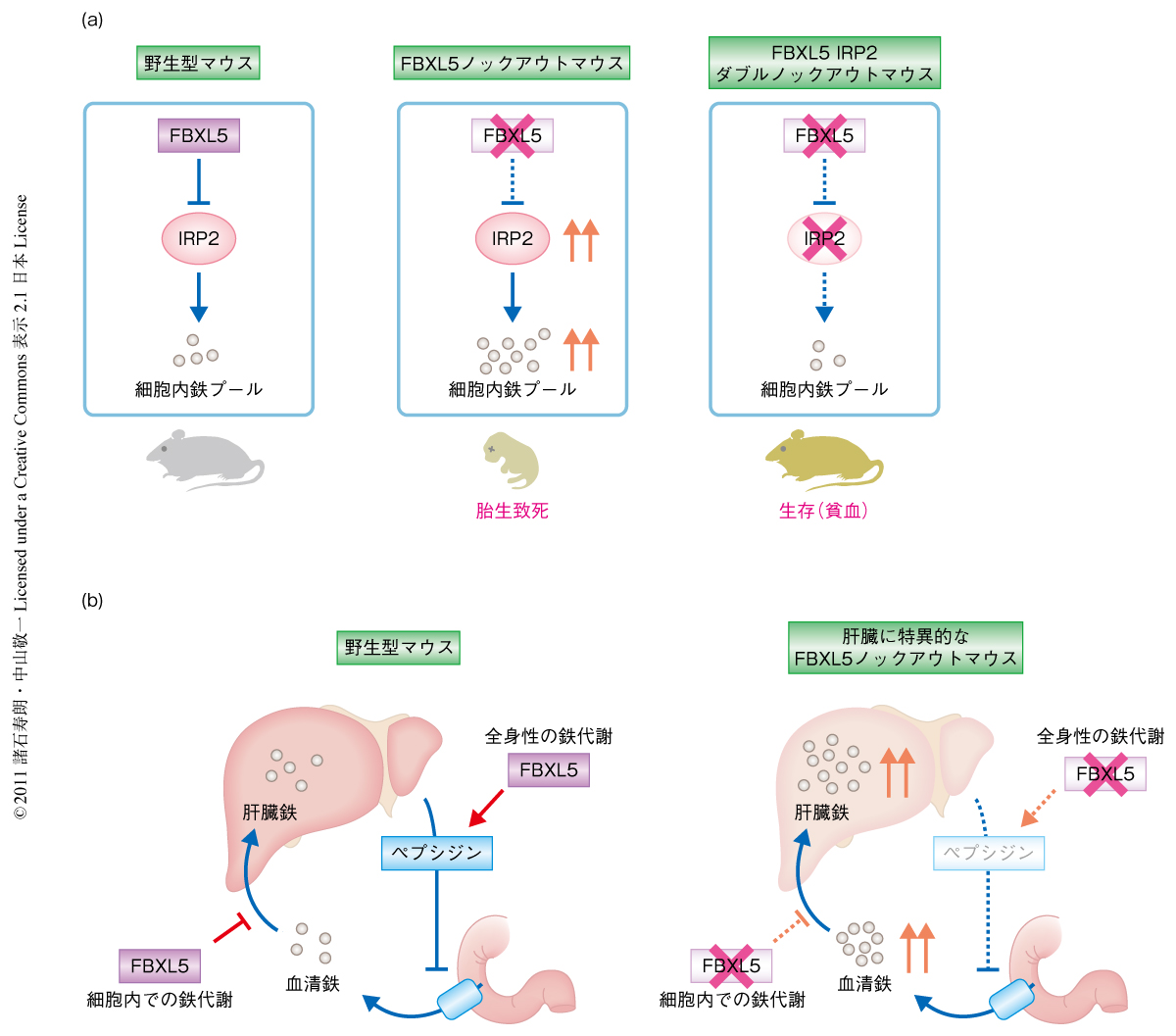
IRP2の蓄積がFBXL5ノックアウトマウスの胎生早期における致死の原因であるという仮説を検証するため,IRP2の追加の欠損がFBXL5ノックアウトマウスの胎生期における致死性を防ぐことができるかどうか検証した.すべてのFBXL5ノックアウトマウスは胎生早期に子宮内で死亡したが,驚いたことに,FBXL5 IRP2ダブルノックアウトマウスはメンデルの法則にしたがった割合で出生し,軽度の小球性貧血を発症するものの,ほぼ正常に発育した.つまり,このFBXL5 IRP2ダブルノックアウトマウスでは,FBXL5ノックアウトマウスにおけるIRP2の増加が解除され,鉄の過剰な蓄積が阻止されたため胎生期における死亡を回避したものと考えられた(図2a).このことは,FBXL5ノックアウトマウスの死因がIRP2の過剰な活性化にあることを遺伝学的に証明しており,FBXL5とIRP2による細胞における鉄の量の管理が生体における鉄代謝の中心的な制御機構であることを示していた.
成体マウスにおけるFBXL5の機能を調べるため肝細胞に特異的にFbxl5遺伝子を欠損させた.この肝臓特異的FBXL5ノックアウトマウスは生存可能であったが,肝臓ではIRP2が過剰に活性化しておりFe2+が蓄積していた.また,肝臓の組織病理学的な解析では肝臓の炎症を示す炎症性細胞の小葉性浸潤が観察された.さらに,脂肪染色を行うと肝臓において核の偏在化をともなわない複数の小さな脂肪滴の沈着を認め,ミトコンドリアの機能障害に関連する肝臓の障害の特性5) を示した.このミトコンドリアの機能障害の原因を探るため遠心分画法を用いて細胞のどこに鉄が蓄積しているのかを調べたところ,FBXL5を欠損した細胞ではミトコンドリアにおいて鉄の量が増加していた.FBXL5を欠損した細胞ではミトコンドリア機能を示すATP産生量が減少し,また,肝臓特異的FBXL5ノックアウトマウスの肝臓を電子顕微鏡で観察するとミトコンドリアの損傷を示唆する膨大化した低濃度のミトコンドリアが観察された.これらの結果から,肝臓特異的FBXL5ノックアウトマウスでは肝臓においてIRP2が過剰に活性化するため,鉄の主要な利用器官であるミトコンドリアへの鉄の供給が増加し,ミトコンドリアの機能障害から脂肪肝炎を発症することが示唆された.
FBXL5の肝臓特異的な欠損が全身性の鉄恒常性にどのような影響をあたえるか検討した.肝臓は個体レベルでの鉄代謝の制御を担う中心臓器であり,鉄の過剰時にペプチドホルモンであるヘプシジンを産生することで腸管からの鉄の吸収を抑制し血中における鉄の過剰な増加を抑制している6).ところが,肝臓特異的FBXL5ノックアウトマウスではヘプシジンの産生が低下しており,その結果,血清における鉄濃度およびトランスフェリン飽和度が著しく増加していた.このことは,肝臓特異的なFBXL5の欠損が肝臓からのヘプシジンの分泌の低下をひき起こし,全身性の鉄過剰をきたすことを示唆していた(図2b).
そこで,肝臓特異的FBXL5ノックアウトマウスを高鉄含有食で飼育することで鉄の過剰負荷があたえる影響を調べた.すると,対照マウスはすべて生き残ったのに対し,ほとんどの肝臓特異的FBXL5ノックアウトマウスは高鉄含有食の投与ののち1日以内に死亡した.高鉄含有食の投与後1日の肝臓を解析すると,肝臓特異的FBXL5ノックアウトマウスでは脂肪肝の表面に大量の出血が認められた.アスパラギン酸アミノトランスフェラーゼ,アラニンアミノトランスフェラーゼ,乳酸脱水素酵素など肝臓に発現する酵素の血清におけるレベルは肝臓特異的FBXL5ノックアウトマウスにおいて約100倍にも上昇しており,肝細胞の急性進行性の破壊の起こっていることが示唆された.組織病理学的な解析では,FBXL5を欠損した肝臓においてFe2+の過剰な蓄積とそれにともなう酸化ストレスから肝細胞がアポトーシスをきたし,おもに門脈の周辺に大規模な細胞死の起こっていることが明らかになった.さらに,肝臓特異的FBXL5ノックアウトマウスの肝臓における酸化ストレスと細胞死は,高鉄含有食による飼育の際に飲料水へ抗酸化作用のあるN-アセチル-L-システインを添加することにより顕著に減衰した.これらの結果から,正常な成体の肝臓において,FBXL5は肝細胞の鉄代謝と全身性の鉄代謝の両方を制御することで鉄過剰による傷害から肝臓を守る防御機構としてはたらいており,この防御機構のなくなったマウスでは高鉄含有食による飼育において鉄の過剰に起因する急性肝不全により死亡してしまうことが明らかになった(図2b).
筆者らは,FBXL5の欠損はIRP2の発現量の増加と過剰な活性化をまねき,結果として,Fe2+の過剰な蓄積によりマウスの胚または成体の肝臓に致死的な損傷をあたえることを示した.筆者らの示した遺伝学的な証拠は,IRP2がSCF複合体型ユビキチンリガーゼSCFFBXL5の主要な標的であることを示唆した.さらに今回の知見は,FBXL5は細胞における適切な鉄濃度の維持に中心的な役割をはたしており,また,FBXL5が胚発生のためだけでなく,マウスの出生ののちの通常の肝臓の生理にとり必須であることも示した.
近年,肝臓における鉄の過負荷は非アルコール性脂肪性肝炎,C型肝炎ウイルスの感染にともなう慢性肝疾患,および,肝細胞がんの増悪因子となることが注目されている7).過剰なFe2+はフリーラジカルを生成し,酸化ストレスから慢性炎症,DNA損傷,遺伝的な不安定性,および,腫瘍の形成をひき起こす.鉄の蓄積それ自体は慢性肝疾患の発症に寄与しないのかもしれないが,鉄過剰は疾患の進行に有害な影響をあたえる.筆者らは,肝臓特異的なFBXL5の欠損が肝臓における鉄含有量の増加,全身における鉄の過負荷,高鉄含有食による飼育による致死的な急性肝不全をきたすことを示した.肝臓におけるFBXL5の発現低下は鉄の蓄積によりこのような疾患の増悪因子となる可能性があり,今後,FBXL5-IRP2系を制御することで細胞における鉄の量を適切に調節し,これらの疾患に対する治療へ応用されることが期待される.
略歴:九州大学大学院医学系学府博士課程 在学中.
研究テーマ:ユビキチン化による鉄代謝の制御機構.
抱負:生物は知れば知るほど奥が深いと感心します.既存の価値観にとらわれず,しっかりと実態をみつめて一歩ずつ研究を行っていきたいと思います.
中山 敬一(Keiichi I. Nakayama)
九州大学生体防御医学研究所 教授.
研究室URL:http://www.bioreg.kyushu-u.ac.jp/saibou/index.html
© 2011 諸石寿朗・中山敬一 Licensed under CC 表示 2.1 日本
(九州大学生体防御医学研究所 分子医科学分野)
email:諸石寿朗
DOI: 10.7875/first.author.2011.144
The FBXL5-IRP2 axis is integral to control of iron metabolism in vivo.
Toshiro Moroishi, Masaaki Nishiyama, Yukiko Takeda, Kazuhiro Iwai, Keiichi I. Nakayama
Cell Metabolism, 14, 339-351 (2011)
要 約
FBXL5はSCF複合体型ユビキチンリガーゼの基質認識サブユニットであり,細胞において鉄の量が増加した場合に鉄代謝制御タンパク質IRP2の分解を促進すると考えられているが,生体における鉄代謝の制御にFBXL5のはたす役割はほとんど理解されていない.筆者らは,マウスの全身においてFBXL5を欠損させるとIRP2の過剰な活性化が起こり,鉄の蓄積により胎生早期に大規模なアポトーシスを起こして致死となることを発見した.この胎生期における致死性はIRP2との二重欠損により回避されたため,FBXL5を欠損したマウスのおもな死因はIRP2の過剰な活性化に起因するものと考えられた.また,肝臓に特異的にFBXL5を欠損させると生まれたマウスは鉄過剰症による脂肪性肝炎を発症し,さらに,高鉄含有食をあたえると1日で急性肝不全を発症し死亡した.以上の結果から,細胞への鉄の適切な供給を確保するというFBXL5の主要な役割が明らかになった.
はじめに
鉄は電子の授受を容易に行いうることから種々の酵素の活性中心としてはたらいており,地球上のほぼすべての生物にとりその生存に必須な元素である.一方で,過剰に二価鉄(Fe2+)が存在すると,その高い反応性ゆえにフリーラジカルの産生を促進し細胞に対する傷害性をもたらす.つまり,鉄は不足しても過剰でも生体に悪影響を及ぼすため,生体においては鉄の量がつねに適切な量になるよう厳密に調節される必要がある.
細胞における鉄恒常性は,鉄の取り込み,利用,貯蔵,および,排出の協調的な制御により維持されている1)(図1).鉄はFe2+のかたちで細胞に入り,タンパク質への直接的な取り込みや,鉄の利用の主要な器官であるミトコンドリアへの供給に必要な細胞質鉄プールとして蓄積される.利用されない細胞質鉄プールのFe2+は,鉄を介した細胞の損傷を防ぐためフェリチンと結合して酸化還元反応を起こさない三価鉄(Fe3+)として貯蔵されるか,あるいは,フェロポーチンにより細胞外に排出される.
これらの鉄代謝関連タンパク質の発現量はIRP2(iron-regulatory protein 2)により協調的に制御されている2).IRP2はmRNA結合タンパク質であり,鉄の欠乏条件においてフェリチンやフェロポーチン,または,Alas2(aminolevulinic acid synthase 2)のmRNAの5’側非翻訳領域に結合して翻訳の開始を阻害するとともに,トランスフェリン受容体1のmRNAの3’側非翻訳領域に結合してその分解を防ぐ.これらの相互作用により,IRP2は鉄の欠乏条件においてフェリチン(鉄の貯蔵を行う)やフェロポーチン(鉄の排出を行う),Alas2(鉄の利用を行う)の発現量を低下させ,逆に,トランスフェリン受容体1(鉄の取り込みを行う)の発現量を増加させることで,結果として細胞質鉄プールのサイズを大きくする.一方,鉄の過剰条件ではIRP2はSCF複合体型ユビキチンリガーゼSCFFBXL5により分解される3,4).SCFFBXL5の基質認識サブユニットFBXL5(F-box and leucine-rich repeat protein 5)は鉄の欠乏時には不安定であるが,過剰時には自らのもつ鉄結合ドメインへの鉄の配位により安定化しIRP2を分解できるようになる.このように,FBXL5は鉄の過剰条件でのIRP2の分解において不可欠な役割をもつにもかかわらず,生体における鉄恒常性の維持にFBXL5のはたす役割はまったく明らかになっていなかった.今回,筆者らは,FBXL5-IRP2系が生体における鉄恒常性の維持の中心的な制御機構であることを明らかにした.
1.FBXL5ノックアウトマウスは鉄の過剰な蓄積により胎生早期に死亡する
鉄代謝の制御におけるFBXL5の機能を解析するため,全身においてFbxl5遺伝子を欠損させたFBXL5ノックアウトマウスを作製した.FBXL5ノックアウトマウスの胚は胎生7.5日までは正常に発育したが,胎生8.5日以降には発育遅延と大規模な出血を示し死亡した.組織病理学的な解析を行うと,FBXL5ノックアウトマウスの胚では早期胎盤領域においてFe2+の過剰な蓄積とそれにともなう酸化傷害を認め,凝縮核をともなうアポトーシス細胞が観察された.つぎに,胎盤の機能とは無関係にFBXL5ノックアウトマウスの胚の成長能力を直接に評価するため,胚盤胞をin vitroで培養しその成長を調べた.通常の鉄条件において培養すると,野生型マウスおよびFBXL5ノックアウトマウスの胚盤胞はともに正常に発育した.しかしながら,鉄の過剰条件において培養すると,野生型マウスの胚盤胞は正常に発育したのに対し,FBXL5ノックアウトマウスの胚盤胞の発育は障害された.そして,この発育障害は抗酸化作用のあるN-アセチル-L-システインの投与により回避された.これらの結果から,FBXL5ノックアウトマウスでは鉄過剰ストレスに対する防御機構が破綻しており,Fe2+の過剰な蓄積にともなう酸化ストレスにより傷害をうけていることがわかった.
FBXL5ノックアウトマウスの胚において鉄の過剰な蓄積が観察されたため,IRP2の異常な活性化が鉄恒常性の破綻の原因であるかもしれないという仮説をたてた.実際,FBXL5ノックアウトマウスの胚ではIRP2の下流の標的であるトランスフェリン受容体1のmRNA量が著しく増加しており,IRP2の活性の増加していることが示唆された.FBXL5ノックアウトマウスは胎生早期に致死となるため,鉄恒常性の維持におけるFBXL5の役割を詳細に解析することはむずかしかった.そこで,Fbxl5遺伝子を組織特異的に欠損するようなコンディショナルノックアウトマウスを作製した.このノックアウトマウスから胎仔線維芽細胞を調製しFbxl5遺伝子を欠損させたところ,IRP2の鉄濃度に依存的な分解が障害され,通常はIRP2が分解されるべき鉄過剰の条件においてもIRP2が蓄積していた.この結果と一致して,IRP2の標的となるmRNAのコードするタンパク質の発現量もまたその制御に異常をきたしていた.すなわち,トランスフェリン受容体1の発現量は増加しており,一方,フェリチンの発現量は減少していた.これらの結果は,FBXL5を欠損したマウス胎仔または胎仔線維芽細胞では,細胞における鉄の状態とは無関係にIRP2が蓄積した結果,IRP2の標的の制御に異常をきたしていることを示唆していた(図1).つまり,FBXL5ノックアウトマウスではIRP2が過剰に活性化し,細胞質鉄プールの増大からFe2+の過剰な蓄積とそれにともなう酸化ストレスをきたし,胎生期に死亡することが示唆された.
2.FBXL5とIRP2とのダブルノックアウトマウスは正常に発育する
IRP2の蓄積がFBXL5ノックアウトマウスの胎生早期における致死の原因であるという仮説を検証するため,IRP2の追加の欠損がFBXL5ノックアウトマウスの胎生期における致死性を防ぐことができるかどうか検証した.すべてのFBXL5ノックアウトマウスは胎生早期に子宮内で死亡したが,驚いたことに,FBXL5 IRP2ダブルノックアウトマウスはメンデルの法則にしたがった割合で出生し,軽度の小球性貧血を発症するものの,ほぼ正常に発育した.つまり,このFBXL5 IRP2ダブルノックアウトマウスでは,FBXL5ノックアウトマウスにおけるIRP2の増加が解除され,鉄の過剰な蓄積が阻止されたため胎生期における死亡を回避したものと考えられた(図2a).このことは,FBXL5ノックアウトマウスの死因がIRP2の過剰な活性化にあることを遺伝学的に証明しており,FBXL5とIRP2による細胞における鉄の量の管理が生体における鉄代謝の中心的な制御機構であることを示していた.
3.肝臓に特異的なFBXL5ノックアウトマウスは脂肪肝炎を発症する
成体マウスにおけるFBXL5の機能を調べるため肝細胞に特異的にFbxl5遺伝子を欠損させた.この肝臓特異的FBXL5ノックアウトマウスは生存可能であったが,肝臓ではIRP2が過剰に活性化しておりFe2+が蓄積していた.また,肝臓の組織病理学的な解析では肝臓の炎症を示す炎症性細胞の小葉性浸潤が観察された.さらに,脂肪染色を行うと肝臓において核の偏在化をともなわない複数の小さな脂肪滴の沈着を認め,ミトコンドリアの機能障害に関連する肝臓の障害の特性5) を示した.このミトコンドリアの機能障害の原因を探るため遠心分画法を用いて細胞のどこに鉄が蓄積しているのかを調べたところ,FBXL5を欠損した細胞ではミトコンドリアにおいて鉄の量が増加していた.FBXL5を欠損した細胞ではミトコンドリア機能を示すATP産生量が減少し,また,肝臓特異的FBXL5ノックアウトマウスの肝臓を電子顕微鏡で観察するとミトコンドリアの損傷を示唆する膨大化した低濃度のミトコンドリアが観察された.これらの結果から,肝臓特異的FBXL5ノックアウトマウスでは肝臓においてIRP2が過剰に活性化するため,鉄の主要な利用器官であるミトコンドリアへの鉄の供給が増加し,ミトコンドリアの機能障害から脂肪肝炎を発症することが示唆された.
4.肝臓に特異的なFBXL5ノックアウトマウスは高鉄含有食による飼育により急性肝不全を発症し死亡する
FBXL5の肝臓特異的な欠損が全身性の鉄恒常性にどのような影響をあたえるか検討した.肝臓は個体レベルでの鉄代謝の制御を担う中心臓器であり,鉄の過剰時にペプチドホルモンであるヘプシジンを産生することで腸管からの鉄の吸収を抑制し血中における鉄の過剰な増加を抑制している6).ところが,肝臓特異的FBXL5ノックアウトマウスではヘプシジンの産生が低下しており,その結果,血清における鉄濃度およびトランスフェリン飽和度が著しく増加していた.このことは,肝臓特異的なFBXL5の欠損が肝臓からのヘプシジンの分泌の低下をひき起こし,全身性の鉄過剰をきたすことを示唆していた(図2b).
そこで,肝臓特異的FBXL5ノックアウトマウスを高鉄含有食で飼育することで鉄の過剰負荷があたえる影響を調べた.すると,対照マウスはすべて生き残ったのに対し,ほとんどの肝臓特異的FBXL5ノックアウトマウスは高鉄含有食の投与ののち1日以内に死亡した.高鉄含有食の投与後1日の肝臓を解析すると,肝臓特異的FBXL5ノックアウトマウスでは脂肪肝の表面に大量の出血が認められた.アスパラギン酸アミノトランスフェラーゼ,アラニンアミノトランスフェラーゼ,乳酸脱水素酵素など肝臓に発現する酵素の血清におけるレベルは肝臓特異的FBXL5ノックアウトマウスにおいて約100倍にも上昇しており,肝細胞の急性進行性の破壊の起こっていることが示唆された.組織病理学的な解析では,FBXL5を欠損した肝臓においてFe2+の過剰な蓄積とそれにともなう酸化ストレスから肝細胞がアポトーシスをきたし,おもに門脈の周辺に大規模な細胞死の起こっていることが明らかになった.さらに,肝臓特異的FBXL5ノックアウトマウスの肝臓における酸化ストレスと細胞死は,高鉄含有食による飼育の際に飲料水へ抗酸化作用のあるN-アセチル-L-システインを添加することにより顕著に減衰した.これらの結果から,正常な成体の肝臓において,FBXL5は肝細胞の鉄代謝と全身性の鉄代謝の両方を制御することで鉄過剰による傷害から肝臓を守る防御機構としてはたらいており,この防御機構のなくなったマウスでは高鉄含有食による飼育において鉄の過剰に起因する急性肝不全により死亡してしまうことが明らかになった(図2b).
おわりに
筆者らは,FBXL5の欠損はIRP2の発現量の増加と過剰な活性化をまねき,結果として,Fe2+の過剰な蓄積によりマウスの胚または成体の肝臓に致死的な損傷をあたえることを示した.筆者らの示した遺伝学的な証拠は,IRP2がSCF複合体型ユビキチンリガーゼSCFFBXL5の主要な標的であることを示唆した.さらに今回の知見は,FBXL5は細胞における適切な鉄濃度の維持に中心的な役割をはたしており,また,FBXL5が胚発生のためだけでなく,マウスの出生ののちの通常の肝臓の生理にとり必須であることも示した.
近年,肝臓における鉄の過負荷は非アルコール性脂肪性肝炎,C型肝炎ウイルスの感染にともなう慢性肝疾患,および,肝細胞がんの増悪因子となることが注目されている7).過剰なFe2+はフリーラジカルを生成し,酸化ストレスから慢性炎症,DNA損傷,遺伝的な不安定性,および,腫瘍の形成をひき起こす.鉄の蓄積それ自体は慢性肝疾患の発症に寄与しないのかもしれないが,鉄過剰は疾患の進行に有害な影響をあたえる.筆者らは,肝臓特異的なFBXL5の欠損が肝臓における鉄含有量の増加,全身における鉄の過負荷,高鉄含有食による飼育による致死的な急性肝不全をきたすことを示した.肝臓におけるFBXL5の発現低下は鉄の蓄積によりこのような疾患の増悪因子となる可能性があり,今後,FBXL5-IRP2系を制御することで細胞における鉄の量を適切に調節し,これらの疾患に対する治療へ応用されることが期待される.
文 献
- Andrews, N. C. & Schmidt, P. J.: Iron homeostasis. Annu. Rev. Physiol., 69, 69-85 (2007)[PubMed]
- Muckenthaler, M. U., Galy, B. & Hentze, M. W.: Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr., 28, 197-213 (2008)[PubMed]
- Vashisht, A. A., Zumbrennen, K. B., Huang, X. et al.: Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science, 326, 718-721 (2009)[PubMed]
- Salahudeen, A. A., Thompson, J. W., Ruiz, J. C. et al.: An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science, 326, 722-726 (2009)[PubMed]
- Burt, A. D.: Steatosis and steatohepatitis. Curr. Diagn. Pathol., 7, 141-147 (2001)
- Hentze, M. W., Muckenthaler, M. U., Galy, B. et al.: Two to tango: regulation of Mammalian iron metabolism. Cell, 142, 24-38 (2010)[PubMed]
- Sorrentino, P., D’Angelo, S., Ferbo, U. et al.: Liver iron excess in patients with hepatocellular carcinoma developed on non-alcoholic steato-hepatitis. J. Hepatol., 50, 351-357 (2009)[PubMed]
著者プロフィール
略歴:九州大学大学院医学系学府博士課程 在学中.
研究テーマ:ユビキチン化による鉄代謝の制御機構.
抱負:生物は知れば知るほど奥が深いと感心します.既存の価値観にとらわれず,しっかりと実態をみつめて一歩ずつ研究を行っていきたいと思います.
中山 敬一(Keiichi I. Nakayama)
九州大学生体防御医学研究所 教授.
研究室URL:http://www.bioreg.kyushu-u.ac.jp/saibou/index.html
© 2011 諸石寿朗・中山敬一 Licensed under CC 表示 2.1 日本
