p57は造血幹細胞の静止期の維持および幹細胞性の維持に重要な役割を担う
松本有樹修・中山敬一
(九州大学生体防御医学研究所 分子医科学分野)
email:松本有樹修
DOI: 10.7875/first.author.2011.138
p57 is required for quiescence and maintenance of adult hematopoietic stem cells.
Akinobu Matsumoto, Shoichiro Takeishi, Tomoharu Kanie, Etsuo Susaki, Ichiro Onoyama, Yuki Tateishi, Keiko Nakayama, Keiichi I. Nakayama
Cell Stem Cell, 9, 262-271 (2011)
造血幹細胞はほとんど増殖しておらず,その幹細胞性の維持には静止期にとどまっていることが重要であると考えられている.静止期の維持を担う主要なタンパク質として,CDK阻害タンパク質p21,p27,p57が知られている.しかし,正常の条件ではp21とp27のどちらも造血幹細胞の静止期の維持および幹細胞性の維持には寄与していないことが知られていた一方,造血幹細胞におけるp57の役割は未解明のままであった.今回,筆者らは,造血幹細胞においてp57を欠損したマウスを作製し解析したところ,対照となるマウスと比較してp57を欠損したマウスでは,造血幹細胞数の減少,静止期の割合の減少,造血幹細胞の幹細胞としての機能の低下,などが確認された.これらの結果より,p21でもp27でもなく,p57が造血幹細胞の静止期の維持および幹細胞性の維持に重要なタンパク質であることが明らかになった.
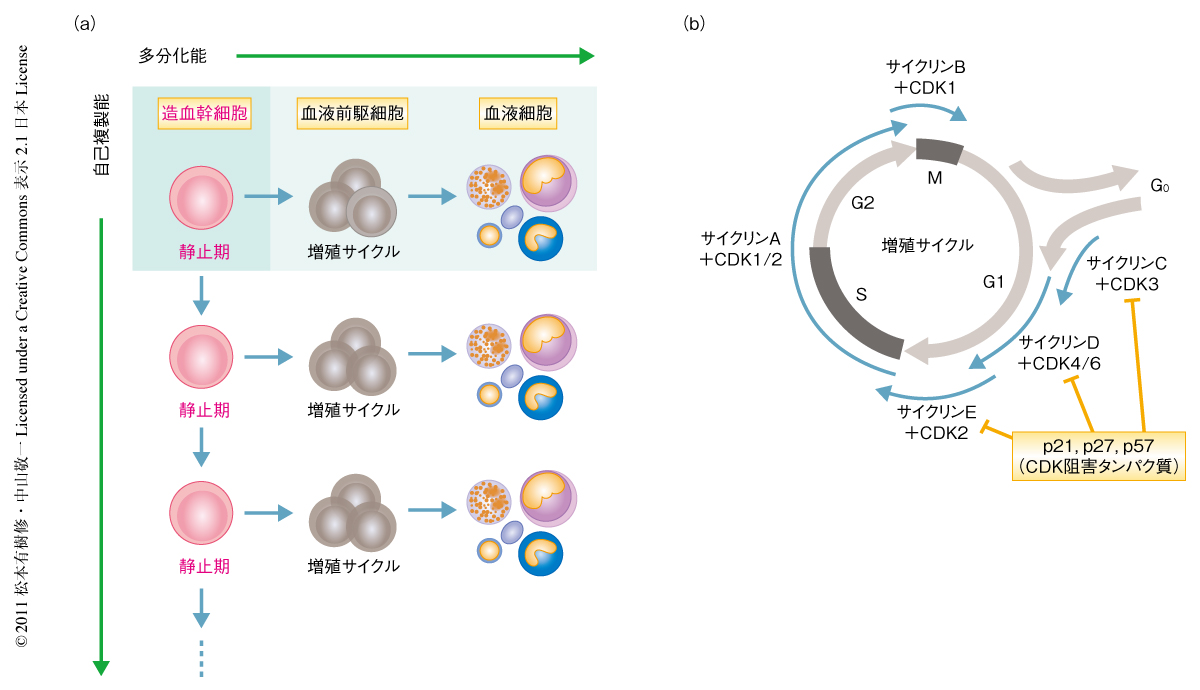
血液には白血球や赤血球,血小板などといった非常に多彩な細胞が存在する.これらすべての細胞は造血幹細胞という共通の細胞からつくられている.造血幹細胞は細胞増殖をほとんどしておらず,細胞周期の静止期にとどまっている(低増殖性).しかし,ごくたまに細胞分裂し,そのときには自己を複製する場合(自己複製能)と,血液前駆細胞をへて多くの血液細胞を産生する場合(多分化能)とがある(図1a).この自己複製能と多分化能とをバランスよく保つことが幹細胞の維持に重要だと考えられている1).造血幹細胞から生み出された血液前駆細胞は活発に細胞増殖し,そののち,さまざまな血液細胞へと分化していく.つまり,造血幹細胞はたまにしか増殖しないが,血液前駆細胞はさかんに増殖し血液全体のバランスを保っている.しかし,なぜ造血幹細胞はあまり分裂しないのか,さらに,分裂しないことが本当に幹細胞の維持に重要であるのかについては,いままでよくわかっていなかった.
細胞分裂を抑制するタンパク質として,3種類のCDK阻害タンパク質,p21,p27,p57が知られている(CDK:cyclin-dependent kinase,サイクリン依存性キナーゼ).これまでの解析により,正常の条件ではp21とp27は造血幹細胞の増殖制御に寄与していないことが知られていた.一方,p57の造血幹細胞における役割はいまだ不明のままであった.今回,筆者らは,造血幹細胞においてp57を欠損したマウスを作製し,p57が造血幹細胞における静止期の維持および幹細胞性の維持に必須のタンパク質であることを明らかにした.
細胞が増殖するためには,サイクリン-CDK複合体が活性化することが必須である.CDK阻害タンパク質とよばれるp21,p27,p57は,サイクリン-CDK複合体の機能を阻害することにより細胞の増殖サイクルを抑制していることが知られている2)(図1b).
これまで,p21ノックアウトマウスあるいはp27ノックアウトマウスを用いることにより造血幹細胞におけるp21やp27の機能解析が行われている.p21はDNA損傷などのストレスの際に発現が上昇し造血幹細胞の維持に寄与しているが,正常のときの造血幹細胞の機能には必須でないことが示されていた3,4).また,p27は造血幹細胞の機能維持に必須でないことが知られていた5).一方,造血幹細胞におけるp57の機能はこれまでまったく解析されていなかった.そのおもな理由は,p57ノックアウトマウスは出生の直後に死亡してしまうため解析が困難であることにあった6).つまり,造血幹細胞は発生期にさかんに増殖し生後になると増殖を停止するため,造血幹細胞の細胞周期の停止機構についてはマウスの生後に解析する必要があるのだが,出生の直後に死亡してしまう従来のp57ノックアウトマウスでは生後の造血幹細胞の機能解析はできなかったのである.
さらに,p57は胎生期には多くの組織において高発現しているが,生後になると大部分の細胞においてほとんど消失してしまう.そのため,生後におけるp57の機能はあまり注目されてこなかった.しかし最近になって,生後においてもごくわずかの細胞ではp57が発現していることがわかってきた.たとえば,血液系の細胞においてはp57のmRNAが造血幹細胞に高発現していることが明らかになった7,8).そこで,タンパク質レベルにおいて血液系の細胞におけるCDK阻害タンパク質の発現を確認してみたところ,やはりp57は造血幹細胞において高発現していた.一方,p21はどの細胞画分においても発現を確認することができず,p27はどの細胞画分においても発現していた.これらの結果は,p57が造血幹細胞の静止期の維持に重要である可能性を強く示唆していた.
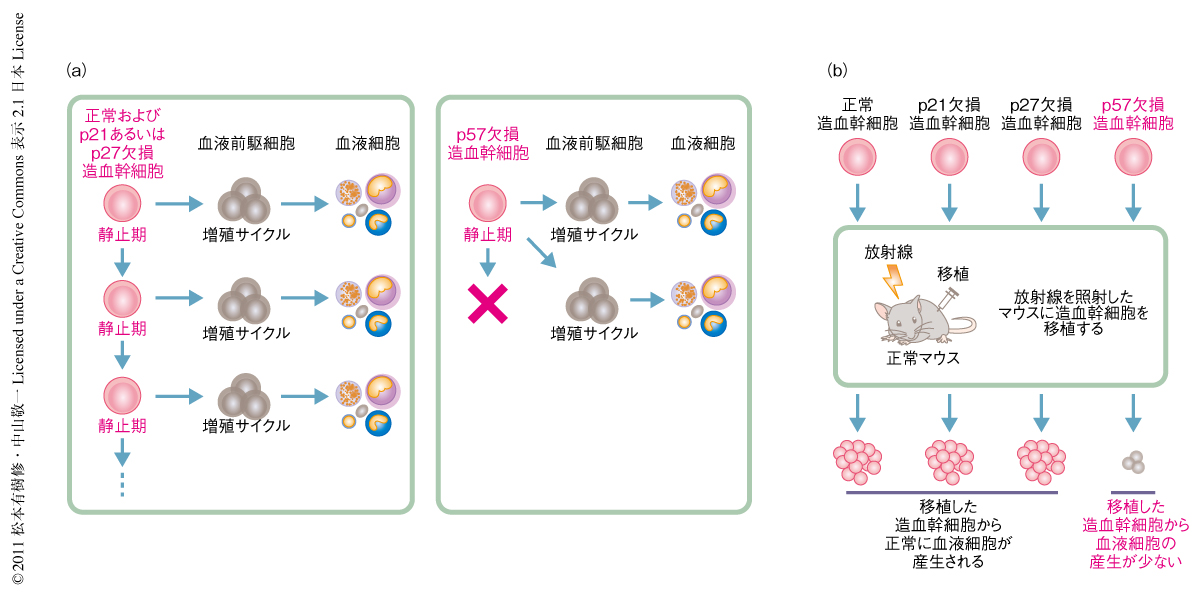
そこで,生後のマウスの造血幹細胞におけるp57の機能解析を行うことにした.p57ノックアウトマウスは出生の直後に死亡してしまうため,血液細胞に特異的にp57を欠損するコンディショナルノックアウトマウスを作製した.対照としてp21ノックアウトマウスやp27ノックアウトマウスも同時に解析した.まず,これらの変異マウスで造血幹細胞の増殖能に変化がないか実験を行った.以前の報告どおり,造血幹細胞からp21あるいはp27が欠損しても造血幹細胞の増殖の割合に変化はなかったが,p57が欠損するとその増殖が異常に亢進することがわかった.しかし,この異常な増殖亢進にもかかわらず,時間の経過ともに造血幹細胞の数は減少していった.これは,造血幹細胞においてp57がなくなると,増殖性が亢進することにより自己を複製せずに血液前駆細胞への分化の方向へかたむいてしまい,その結果として,やがて造血幹細胞が枯渇していくことが考えられた(図2a).一方,p21あるいはp27が欠損しても造血幹細胞数の減少することはなかった.
p57を介したどのような分子機構が造血幹細胞の静止期の維持に重要であるかを検討した.一般的に,p57などのCDK阻害タンパク質はサイクリン-CDK複合体の機能を阻害することが知られている(図1b).活性型のサイクリン-CDK複合体はRbタンパク質をリン酸化することにより細胞周期を活性化する2).そこで,造血幹細胞においてp57が欠損するとサイクリン-CDK複合体の異常な活性化によるRbのリン酸化の異常な亢進が生じるのではないかと考えた.予想どおり,p57を欠損した造血幹細胞に特異的にRbのリン酸化の異常な亢進が起こっており,一方,血液前駆細胞などではRbのリン酸化の亢進は確認されなかった.さらに,p57を欠損した造血幹細胞のサイクリン-CDK複合体の活性を薬剤により阻害することで,造血幹細胞数の減少は抑制された.これらのことより,p57はサイクリン-CDK複合体の機能を抑制することにより,造血幹細胞の静止期を維持していることが明らかになった.
造血幹細胞が血液細胞を産生する能力を生体において測定するため,造血幹細胞の移植実験を行った.p21,p27,p57をそれぞれ欠損したマウスから造血幹細胞を取り出し,放射線を照射した別のマウスに移植した.移植された造血幹細胞は,マウスの体内において自己を複製することで造血幹細胞を十分に増やしたのち血液細胞を産生する.p21あるいはp27を欠損した造血幹細胞は正常の造血幹細胞と同じ程度に血液細胞を産生することができた.一方,p57を欠損した造血幹細胞は正常の造血幹細胞の1/10程度しか血液細胞を産生できなかった(図2b).p57を欠損した造血幹細胞は,自己を複製せずに血液前駆細胞を異常に産生してしまうため,造血幹細胞を増やすことができずに枯渇してしまい,その結果,最終的に産生できる血液細胞の量も非常に少なくなってしまったものと考えられた.
以上の結果より,p57は造血幹細胞に多く存在し,造血幹細胞の増殖速度が速くなりすぎないよう維持していることが明らかになった.また,p57がなくなった造血幹細胞は異常に血液前駆細胞を産生して自己を複製できなくなり,最終的には造血幹細胞は枯渇してしまった.これらのことより,造血幹細胞がその機能を維持するためには,やはり長期間にわたり静止期にとどまっていることが重要であることも明らかになった.この研究により,造血幹細胞の自己複製機構の一端が明らかになった.
現在,輸血は他人の血液をもらうという方法で行われているが,これには供給量や感染事故などの問題があり,いずれ自分の造血幹細胞から試験管内において大量に血液を産生して自分にもどす“自己産生輸血”という方式に切り替わることが期待されている.しかし,造血幹細胞は体外に取り出すとストレスにより自己を複製できず枯渇してしまうため,現在では体外において十分な量の血液を得ることは不可能である.この問題を克服するためには,造血幹細胞の自己複製機構を理解し,それを適切に制御する技術を確立することが必要である.今回の筆者らの解析により,p57により造血幹細胞が適切な増殖速度を保つことがその自己複製に重要であることが明らかになった.この分子機構を詳細に調べることにより,将来的には機能を喪失することなく大量に造血幹細胞を殖やす技術が確立されると期待される.試験管内での造血幹細胞の大量複製が可能となれば,輸血だけでなく,白血病など多くの血液疾患に対する再生治療への道がおおいに広がることも期待される.
略歴:2011年 九州大学大学院医学系学府博士課程 修了,同年より同 生体防御医学研究所 学術研究員.
研究テーマ:幹細胞の細胞周期制御.
関心事:long non-coding RNA.
中山 敬一(Keiichi I. Nakayama)
九州大学生体防御医学研究所 教授.
研究室URL:http://www.bioreg.kyushu-u.ac.jp/saibou/index.html
© 2011 松本有樹修・中山敬一 Licensed under CC 表示 2.1 日本
(九州大学生体防御医学研究所 分子医科学分野)
email:松本有樹修
DOI: 10.7875/first.author.2011.138
p57 is required for quiescence and maintenance of adult hematopoietic stem cells.
Akinobu Matsumoto, Shoichiro Takeishi, Tomoharu Kanie, Etsuo Susaki, Ichiro Onoyama, Yuki Tateishi, Keiko Nakayama, Keiichi I. Nakayama
Cell Stem Cell, 9, 262-271 (2011)
要 約
造血幹細胞はほとんど増殖しておらず,その幹細胞性の維持には静止期にとどまっていることが重要であると考えられている.静止期の維持を担う主要なタンパク質として,CDK阻害タンパク質p21,p27,p57が知られている.しかし,正常の条件ではp21とp27のどちらも造血幹細胞の静止期の維持および幹細胞性の維持には寄与していないことが知られていた一方,造血幹細胞におけるp57の役割は未解明のままであった.今回,筆者らは,造血幹細胞においてp57を欠損したマウスを作製し解析したところ,対照となるマウスと比較してp57を欠損したマウスでは,造血幹細胞数の減少,静止期の割合の減少,造血幹細胞の幹細胞としての機能の低下,などが確認された.これらの結果より,p21でもp27でもなく,p57が造血幹細胞の静止期の維持および幹細胞性の維持に重要なタンパク質であることが明らかになった.
はじめに
血液には白血球や赤血球,血小板などといった非常に多彩な細胞が存在する.これらすべての細胞は造血幹細胞という共通の細胞からつくられている.造血幹細胞は細胞増殖をほとんどしておらず,細胞周期の静止期にとどまっている(低増殖性).しかし,ごくたまに細胞分裂し,そのときには自己を複製する場合(自己複製能)と,血液前駆細胞をへて多くの血液細胞を産生する場合(多分化能)とがある(図1a).この自己複製能と多分化能とをバランスよく保つことが幹細胞の維持に重要だと考えられている1).造血幹細胞から生み出された血液前駆細胞は活発に細胞増殖し,そののち,さまざまな血液細胞へと分化していく.つまり,造血幹細胞はたまにしか増殖しないが,血液前駆細胞はさかんに増殖し血液全体のバランスを保っている.しかし,なぜ造血幹細胞はあまり分裂しないのか,さらに,分裂しないことが本当に幹細胞の維持に重要であるのかについては,いままでよくわかっていなかった.
細胞分裂を抑制するタンパク質として,3種類のCDK阻害タンパク質,p21,p27,p57が知られている(CDK:cyclin-dependent kinase,サイクリン依存性キナーゼ).これまでの解析により,正常の条件ではp21とp27は造血幹細胞の増殖制御に寄与していないことが知られていた.一方,p57の造血幹細胞における役割はいまだ不明のままであった.今回,筆者らは,造血幹細胞においてp57を欠損したマウスを作製し,p57が造血幹細胞における静止期の維持および幹細胞性の維持に必須のタンパク質であることを明らかにした.
1.p57は造血幹細胞において高発現している
細胞が増殖するためには,サイクリン-CDK複合体が活性化することが必須である.CDK阻害タンパク質とよばれるp21,p27,p57は,サイクリン-CDK複合体の機能を阻害することにより細胞の増殖サイクルを抑制していることが知られている2)(図1b).
これまで,p21ノックアウトマウスあるいはp27ノックアウトマウスを用いることにより造血幹細胞におけるp21やp27の機能解析が行われている.p21はDNA損傷などのストレスの際に発現が上昇し造血幹細胞の維持に寄与しているが,正常のときの造血幹細胞の機能には必須でないことが示されていた3,4).また,p27は造血幹細胞の機能維持に必須でないことが知られていた5).一方,造血幹細胞におけるp57の機能はこれまでまったく解析されていなかった.そのおもな理由は,p57ノックアウトマウスは出生の直後に死亡してしまうため解析が困難であることにあった6).つまり,造血幹細胞は発生期にさかんに増殖し生後になると増殖を停止するため,造血幹細胞の細胞周期の停止機構についてはマウスの生後に解析する必要があるのだが,出生の直後に死亡してしまう従来のp57ノックアウトマウスでは生後の造血幹細胞の機能解析はできなかったのである.
さらに,p57は胎生期には多くの組織において高発現しているが,生後になると大部分の細胞においてほとんど消失してしまう.そのため,生後におけるp57の機能はあまり注目されてこなかった.しかし最近になって,生後においてもごくわずかの細胞ではp57が発現していることがわかってきた.たとえば,血液系の細胞においてはp57のmRNAが造血幹細胞に高発現していることが明らかになった7,8).そこで,タンパク質レベルにおいて血液系の細胞におけるCDK阻害タンパク質の発現を確認してみたところ,やはりp57は造血幹細胞において高発現していた.一方,p21はどの細胞画分においても発現を確認することができず,p27はどの細胞画分においても発現していた.これらの結果は,p57が造血幹細胞の静止期の維持に重要である可能性を強く示唆していた.
2.p57は造血幹細胞の静止期の維持に重要である
そこで,生後のマウスの造血幹細胞におけるp57の機能解析を行うことにした.p57ノックアウトマウスは出生の直後に死亡してしまうため,血液細胞に特異的にp57を欠損するコンディショナルノックアウトマウスを作製した.対照としてp21ノックアウトマウスやp27ノックアウトマウスも同時に解析した.まず,これらの変異マウスで造血幹細胞の増殖能に変化がないか実験を行った.以前の報告どおり,造血幹細胞からp21あるいはp27が欠損しても造血幹細胞の増殖の割合に変化はなかったが,p57が欠損するとその増殖が異常に亢進することがわかった.しかし,この異常な増殖亢進にもかかわらず,時間の経過ともに造血幹細胞の数は減少していった.これは,造血幹細胞においてp57がなくなると,増殖性が亢進することにより自己を複製せずに血液前駆細胞への分化の方向へかたむいてしまい,その結果として,やがて造血幹細胞が枯渇していくことが考えられた(図2a).一方,p21あるいはp27が欠損しても造血幹細胞数の減少することはなかった.
3.p57を欠損した造血幹細胞ではサイクリン-CDK複合体の異常な活性化が生じる
p57を介したどのような分子機構が造血幹細胞の静止期の維持に重要であるかを検討した.一般的に,p57などのCDK阻害タンパク質はサイクリン-CDK複合体の機能を阻害することが知られている(図1b).活性型のサイクリン-CDK複合体はRbタンパク質をリン酸化することにより細胞周期を活性化する2).そこで,造血幹細胞においてp57が欠損するとサイクリン-CDK複合体の異常な活性化によるRbのリン酸化の異常な亢進が生じるのではないかと考えた.予想どおり,p57を欠損した造血幹細胞に特異的にRbのリン酸化の異常な亢進が起こっており,一方,血液前駆細胞などではRbのリン酸化の亢進は確認されなかった.さらに,p57を欠損した造血幹細胞のサイクリン-CDK複合体の活性を薬剤により阻害することで,造血幹細胞数の減少は抑制された.これらのことより,p57はサイクリン-CDK複合体の機能を抑制することにより,造血幹細胞の静止期を維持していることが明らかになった.
4.p57を欠損した造血幹細胞では血液細胞を再構築する能力が低下する
造血幹細胞が血液細胞を産生する能力を生体において測定するため,造血幹細胞の移植実験を行った.p21,p27,p57をそれぞれ欠損したマウスから造血幹細胞を取り出し,放射線を照射した別のマウスに移植した.移植された造血幹細胞は,マウスの体内において自己を複製することで造血幹細胞を十分に増やしたのち血液細胞を産生する.p21あるいはp27を欠損した造血幹細胞は正常の造血幹細胞と同じ程度に血液細胞を産生することができた.一方,p57を欠損した造血幹細胞は正常の造血幹細胞の1/10程度しか血液細胞を産生できなかった(図2b).p57を欠損した造血幹細胞は,自己を複製せずに血液前駆細胞を異常に産生してしまうため,造血幹細胞を増やすことができずに枯渇してしまい,その結果,最終的に産生できる血液細胞の量も非常に少なくなってしまったものと考えられた.
おわりに
以上の結果より,p57は造血幹細胞に多く存在し,造血幹細胞の増殖速度が速くなりすぎないよう維持していることが明らかになった.また,p57がなくなった造血幹細胞は異常に血液前駆細胞を産生して自己を複製できなくなり,最終的には造血幹細胞は枯渇してしまった.これらのことより,造血幹細胞がその機能を維持するためには,やはり長期間にわたり静止期にとどまっていることが重要であることも明らかになった.この研究により,造血幹細胞の自己複製機構の一端が明らかになった.
現在,輸血は他人の血液をもらうという方法で行われているが,これには供給量や感染事故などの問題があり,いずれ自分の造血幹細胞から試験管内において大量に血液を産生して自分にもどす“自己産生輸血”という方式に切り替わることが期待されている.しかし,造血幹細胞は体外に取り出すとストレスにより自己を複製できず枯渇してしまうため,現在では体外において十分な量の血液を得ることは不可能である.この問題を克服するためには,造血幹細胞の自己複製機構を理解し,それを適切に制御する技術を確立することが必要である.今回の筆者らの解析により,p57により造血幹細胞が適切な増殖速度を保つことがその自己複製に重要であることが明らかになった.この分子機構を詳細に調べることにより,将来的には機能を喪失することなく大量に造血幹細胞を殖やす技術が確立されると期待される.試験管内での造血幹細胞の大量複製が可能となれば,輸血だけでなく,白血病など多くの血液疾患に対する再生治療への道がおおいに広がることも期待される.
文 献
- Arai, F. & Suda, T.: Maintenance of quiescent hematopoietic stem cells in the osteoblastic niche. Ann. N. Y. Acad. Sci., 1106, 41-53 (2007)[PubMed]
- Sherr, C. J. & Roberts, J. M.: Living with or without cyclins and cyclin-dependent kinases. Genes Dev., 18, 2699-2711 (2004)[PubMed]
- Cheng, T., Rodrigues, N., Shen, H. et al.: Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science, 287, 1804-1808 (2000)[PubMed]
- van Os, R., Kamminga, L. M., Ausema, A. et al.: A limited role for p21Cip1/Waf1 in maintaining normal hematopoietic stem cell functioning. Stem Cells, 25, 836-843 (2007)[PubMed]
- Cheng, T., Rodrigues, N., Dombkowski, D. et al.: Stem cell repopulation efficiency but not pool size is governed by p27kip1. Nat. Med., 6, 1235-1240 (2000)[PubMed]
- Zhang, P., Liegeois, N. J., Wong, C. et al.: Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature, 387, 151-158 (1997)[PubMed]
- Umemoto T., Yamato M., Nishida K. et al.: p57Kip2 is expressed in quiescent mouse bone marrow side population cells. Biochem. Biophys. Res. Commun., 337, 14-21 (2005)[PubMed]
- Yamazaki, S., Iwama, A., Takayanagi, S. I. et al.: Cytokine signals modulated via lipid rafts mimic niche signals and induce hibernation in hematopoietic stem cells. EMBO J., 25, 3515-3523 (2006)[PubMed]
著者プロフィール
略歴:2011年 九州大学大学院医学系学府博士課程 修了,同年より同 生体防御医学研究所 学術研究員.
研究テーマ:幹細胞の細胞周期制御.
関心事:long non-coding RNA.
中山 敬一(Keiichi I. Nakayama)
九州大学生体防御医学研究所 教授.
研究室URL:http://www.bioreg.kyushu-u.ac.jp/saibou/index.html
© 2011 松本有樹修・中山敬一 Licensed under CC 表示 2.1 日本
