ゲートキーパー耐性変異に対しても有効な選択的ALK阻害剤
坂本 洋
(中外製薬鎌倉研究所 創薬研究第二部)
email:坂本 洋
DOI: 10.7875/first.author.2011.088
CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant.
Hiroshi Sakamoto, Toshiyuki Tsukaguchi, Sayuri Hiroshima, Tatsushi Kodama, Takamitsu Kobayashi, Takaaki A. Fukami, Nobuhiro Oikawa, Takuo Tsukuda, Nobuya Ishii, Yuko Aoki
Cancer Cell, 19, 679-690 (2011)
ALKはある種のがんにおいて染色体転座,遺伝子増幅,点変異などの遺伝子異常にともない恒常的に活性化されるチロシンキナーゼである.今回,筆者らは,これまでのキナーゼ阻害剤とは異なる骨格の化学構造をもち経口投与の可能なALK阻害剤CH5424802を同定した.このCH5424802はALKに対し強力かつ選択的なキナーゼ阻害プロファイルを示し,EML4-ALK融合遺伝子を発現する非小細胞肺がんやNPM-ALK融合遺伝子を発現する未分化大細胞型リンパ腫など,ALK遺伝子に異常をもつがん細胞に対しin vitroおよびin vivoにおいて選択的な抗腫瘍活性を示した.さらに,CH5424802はキナーゼ阻害剤に対する共通耐性機構のひとつとして知られるゲートキーパー残基での点変異に対しても有効であった.CH5424802のように耐性変異に対しても有効なALK阻害剤はALK遺伝子変異に依存性のがんをもつ患者に対し有効な治療効果をあたえる可能性が示唆された.
最近のがんの分子標的薬剤はがんに特異的に作用し臨床においても実績をあげており,今後も新たな医薬品の開発が期待される.しかしながら,キナーゼ阻害剤においては,複数のキナーゼを同時に阻害することによる有効治療域の狭さ,あるいは,さまざまな分子機構でひき起こされる薬剤耐性の出現により,治療効果が限定的となってしまうケースがある.したがって,より強力で選択的な阻害剤,あるいは,薬剤耐性をひき起こしにくい薬剤を見い出すことが要求されている.
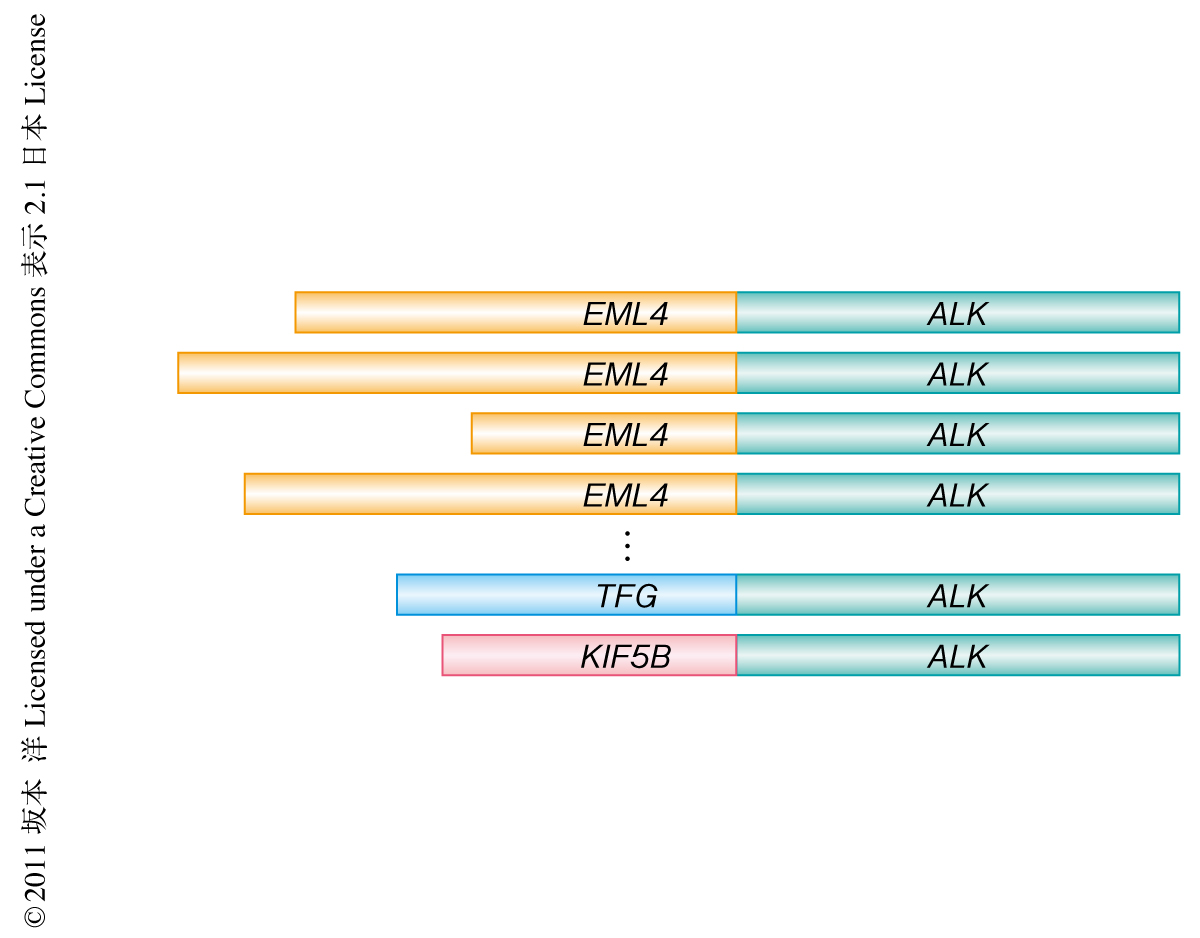
EML4-ALK融合遺伝子は,2007年,非小細胞肺がんにおいて新規に同定された融合がん遺伝子である1).EML4-ALK融合遺伝子の腫瘍形成能は,この融合遺伝子を導入した3T3線維芽細胞を用いた形質転換活性やこの融合遺伝子を肺胞の上皮に特異的に産生するトランスジェニックマウスにおける肺腺がんの発症により確認された1,2).また,EML4-ALK融合遺伝子には融合点の異なる複数のバリアントや3),KIF5B-ALK融合遺伝子のように融合相手の異なる融合遺伝子も同定された4)(図1).このようなALK融合遺伝子をもつ患者は非小細胞肺がん患者に約5%の頻度で認められており,EGF受容体の変異やK-ras遺伝子の変異とは相互に排他的な関係にあることが確認されている5).また,EML4-ALK融合遺伝子をもつ非小細胞肺がん患者はEGF受容体の阻害剤に対する反応に乏しいことから6),これを標的とする新たな治療法の確立が必要とされている.チロシンキナーゼであるc-METおよびALKの阻害剤であるPF-02341066(クリゾチニブ)はALK融合遺伝子をもつ非小細胞肺がん患者を対象にした臨床試験において高い奏効率を示した7).一方で,最近の報告において,クリゾチニブ処置により部分奏効したのち再発の認められた患者のがん検体を用いた遺伝子解析から,ALKにL1196MやC1156Yといった点変異がみつかった8).
今回,筆者らは,ALKに対して高い阻害選択性をもち,かつ,薬剤耐性に寄与する点変異に対しても有効な新しいタイプのALK阻害剤CH5424802を見い出した.
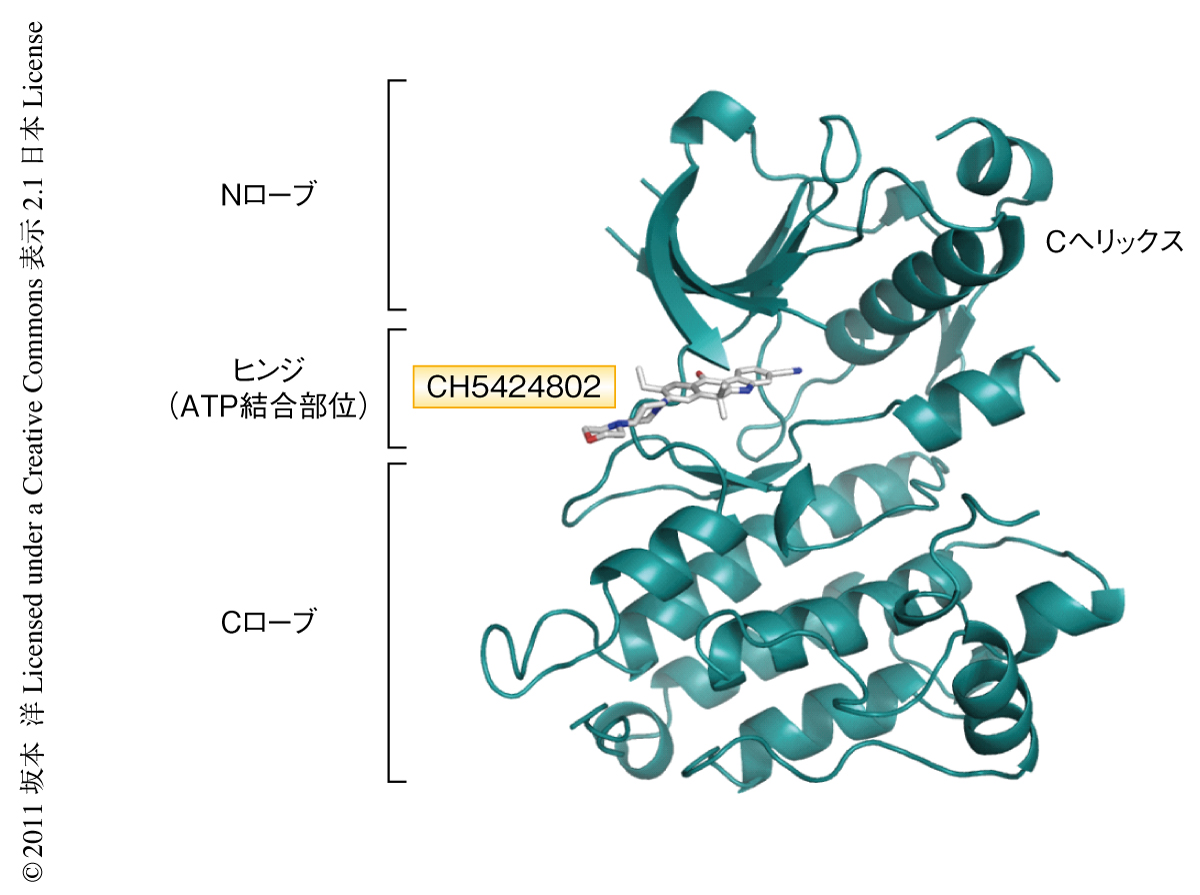
筆者らは,強力かつよりキナーゼ選択性の高いALK阻害剤を取得することに焦点をしぼり化合物の同定を試みた.キナーゼ阻害剤のハイスループットスクリーニングから新規の骨格をもつ複数のヒット化合物が得られ,それらの誘導体のなかでALKを強く阻害するリード化合物をみつけた.このリード化合物の選択性および薬物動態プロファイルを向上するため種々の化学修飾を行い,最終的に経口投与が可能で強力かつALK選択的なベンゾ[b]カルバゾール誘導体CH5424802を同定した.ALKのキナーゼ領域とCH5424802との共結晶のX線構造解析(図2,PDB ID:3AOX),および,生化学的な実験により,CH5424802がALKのATP結合部位においてATPと競合的に結合することを確認した.
in vitroでの酵素阻害活性測定系において,CH5424802のALKに対する50%阻害濃度は1.9 nMであった.また,神経芽腫細胞において活性化型の点変異として同定されているF1174LあるいはR1275Q 9) をもつALKに対しても同じ程度の阻害活性を示すことを確認した.また,CH5424802のキナーゼ選択性を確認するためさまざまなキナーゼに対する阻害活性を測定したところ,c-MET,インスリン受容体,EGF受容体などを含むALK以外の24のキナーゼに対しては弱い阻害活性を示す,あるいは,阻害活性を示さなかった.さらに,細胞を用いた実験において,CH5424802はEML4-ALK融合遺伝子を発現するNCI-H2228細胞においてALKの自己リン酸化を阻害し,それにともないSTAT3およびAKTのリン酸化の抑制も認められた.
チロシンキナーゼであるEGF受容体に対する阻害剤はEGF受容体に変異をもつ患者に対し高い治療効果を示すことが確認され8),がん化に寄与するがん遺伝子産物を標的とした分子標的薬剤の有用性が期待されている.そこで,ALKおよびEGF受容体について遺伝子変異のステータスの異なる非小細胞肺がん細胞株を用い,CH5424802および比較対照としてEGF受容体の阻害剤であるゲフィチニブの抗腫瘍活性を検討した.CH5424802はEML4-ALK融合遺伝子を発現するNCI-H2228細胞において抗腫瘍活性およびアポトーシス誘導活性を示したが,ALK融合遺伝子の発現していない細胞株においてはほとんど抗腫瘍活性を示さなかった.一方,ゲフィチニブはEGF受容体に変異をもつ細胞株に対して高い抗腫瘍活性を示し,NCI-H2228細胞に対する活性は弱かった.これらの結果より,CH5424802がALK融合遺伝子をもつ非小細胞肺がんというセグメントに対し選択的に抗腫瘍活性をもたらす可能性が示された.
同様に,ALK遺伝子に異常をもつ非小細胞肺がん以外のがん細胞株におけるCH5424802の抗腫瘍活性をin vitroでの細胞増殖阻害アッセイにより評価したところ,ALK遺伝子の転座をもつ未分化大細胞型リンパ腫細胞であるKARPAS-299細胞やSR細胞,ALK遺伝子の増幅の認められる神経芽腫細胞であるNB-1細胞,ALK遺伝子に活性化型の点変異F1174LをもつKELLY細胞では,CH5424802に対する高い感受性を示した.一方,CH5424802はALK遺伝子に異常の認められない複数の細胞株では抗腫瘍効果を示さなかった.以上のことから,CH5424802は非小細胞肺がんにかぎらずALK遺伝子に異常をもつさまざまながん細胞に対し選択的に抗腫瘍活性を示すことが明らかになった.
CH5424802のin vivoにおける抗腫瘍効果について細胞株をマウスの皮下に移植したゼノグラフトモデルを用いて評価した.その結果,EML4-ALK融合遺伝子を発現する非小細胞肺がん細胞株NCI-H2228を用いたモデルにおいて用量に依存的な腫瘍増殖の抑制効果と腫瘍の退縮が認められた.この試験におけるマウスの体重減少は認められなかった.一方,ALK融合遺伝子を発現していない非小細胞肺がん細胞株A549を移植したモデルではCH5424802による抗腫瘍効果は認められなかった.NCI-H2228細胞モデルの腫瘍において標的阻害の指標となるALKのリン酸化レベルを測定したところ,CH5424802の投与によりこれが抑制されることも確認した.さらに,やや大きなサイズの腫瘍(340~350 mm3)を用いたNCI-H2228細胞のゼノグラフトモデルにおいて,3週間にわたり60 mg/kgのCH5424802を投与したのち4週間にわたり経過観察したところ,強い抗腫瘍効果は維持され腫瘍の再増殖は認められなかった.また,同様な実験をKARPAS-299細胞やNB-1細胞を用いたゼノグラフトモデルでも評価したところ,腫瘍増殖の抑制効果と腫瘍の退縮が認められた.このように,ALK遺伝子に異常をもつ複数のがん種においてCH5424802のin vivoでの高い抗腫瘍活性が確認された.
ALKのシグナル伝達系に関して,NPM-ALK融合遺伝子を発現する未分化大細胞型リンパ腫においては,その下流のタンパク質として転写因子STAT3が細胞の生存や増殖に重要な役割をはたしていることが知られている10).一方,EML4-ALK融合遺伝子を発現する非小細胞肺がんにおいてはこのような下流のタンパク質についてあまり明確には解析されていない.そこで,CH5424802を処理したのちのNCI-H2228細胞モデルの腫瘍を用いて網羅的な遺伝子解析を行ったところ,複数のSTAT3の標的遺伝子の発現低下が確認された.また,リン酸化レベルの解析において,CH5424802を投与したのちの腫瘍におけるSTAT3のリン酸化の阻害にくわえ,AKTの部分的なリン酸化の抑制も認められた.siRNAを用いた実験において,NPM-ALK融合遺伝子を発現するKARPAS-299細胞ではSTAT3のノックダウンにより十分な細胞の増殖阻害が認められたのに対し,EML4-ALK融合遺伝子を発現するNCI-H2228細胞ではSTAT3のノックダウンは細胞増殖にほとんど影響をあたえなかった.したがって,EML4-ALK融合遺伝子をもつ非小細胞肺がんにおいては,STAT3以外,もしくは,STAT3を含めた複数の下流シグナル経路が細胞増殖に必要である可能性が示唆された.
キナーゼ阻害剤に対する獲得耐性機構のひとつとして,キナーゼドメインにおける点変異が知られている.とりわけ,EGF受容体の点変異T790M,あるいは,BCR-ABL融合タンパク質の点変異T315Iのようなゲートキーパー残基の変異は高い頻度で起こりやすい耐性機構のひとつである.最近の報告により,クリゾチニブの臨床試験において部分奏効したのちに再発の認められた非小細胞肺がん患者のがん検体を用いた遺伝子解析から,EML4-ALK融合タンパク質の点変異L1196M(ゲートキーパー残基における変異)およびC1156Yがみつかっている8).この点変異L1196MをもつALKタンパク質に対するCH5424802の影響を調べたところ,変異をもたないALKと同じ程度の高い酵素阻害活性が認められた.
細胞における影響を試みるため,Ba/F3細胞に野生型のEML4-ALK融合遺伝子および点変異L1196MをもつEML4-ALK融合遺伝子を遺伝子導入した安定細胞株を作製した.ALKに非依存的な親細胞Ba/F3細胞と比較して,野生型あるいは点変異L1196MをもつEML4-ALK融合遺伝子を導入したBa/F3細胞においてはCH5424802に対しいずれも高い感受性を示した.一方,クリゾチニブについては,点変異L1196MをもつEML4-ALK融合遺伝子を導入したBa/F3細胞に対する感受性は親細胞と同じ程度であり,ALKの阻害による影響はほとんど認められなかった.また,この感受性に対する影響は細胞におけるALKのリン酸化レベルの抑制効果と一致していた.さらに,この抗腫瘍活性をin vivoで確認するため,これらの細胞株をマウスに移植したゼノグラフトモデルを用いて評価した.その結果,CH5424802は野生型および点変異L1196MをもつEML4-ALK融合遺伝子のいずれに依存する腫瘍に対しても腫瘍の退縮効果が認められた.
ゲートキーパー耐性変異である点変異L1196Mに対しCH5424802が阻害効果を示した理由を明らかにするため,それらの共結晶のX線構造を用いてCH5424802とALKとの結合様式を確認した.その結果,CH5424802の3位シアノ基とALKのゲートキーパー残基L1196とが効果的なCH/π相互作用をしていることが観察された.また,in silicoモデルの解析により,点変異L1196MをもつALKにおいても同様にこの効果的なCH/π相互作用が維持されていることが示唆された.一方,クリゾチニブについては,L1196残基,および,in silicoモデルにおけるM1196残基との効果的な相互作用は認められなかった.このように,共結晶構造の情報からCH5424802がALKのゲートキーパー耐性変異である点変異L1196Mに対し酵素阻害能を保持できることが考察された.
現在,CH5424802についてはALK融合遺伝子をもつ非小細胞肺がん患者を対象とした第1相/第2相の臨床試験が進行中である.今回,見い出された新規のALK阻害剤CH5424802の特徴のひとつは,ALKに対し高いキナーゼ選択性をもつことである.低分子阻害剤のオフターゲットによる毒性は投与量を上げることのさまたげとなり,最終的に薬効を限定的にしてしまうことがある.成人の正常組織においてALKの発現はほとんど認められておらず,ALKを阻害することによる正常組織への影響は非常に小さいものと考えられるので,より選択的なALK阻害剤は広い治療域を生み出し高い効果が期待される.CH5424802のもうひとつの特徴として,ALK阻害剤に対する耐性機構のひとつであるALKの点変異に対しても有効であることがある.CH5424802は点変異としてL1196Mだけでなく,耐性変異として同様に同定された点変異C1156Yに対しても,in vitroでの酵素阻害,および,点変異C1156YをもつEML4-ALK融合タンパク質に依存した細胞の増殖阻害が確認された.CH5424802はALKに対する阻害が強力でかつ広い有効治療域を兼ね備えていることから,別の新たな点変異により阻害活性がやや落ちたとしても薬効には大きな影響のないものと考えられる.このようなことから,CH5424802はALK阻害剤に対し薬剤耐性をひき起こした患者に対しても効果があるかもしれない.今後,ALK遺伝子の点変異にかぎらず,ALK阻害剤のもつ別の耐性機構(たとえば,ほかのがん遺伝子の遺伝子異常やほかのタンパク質の活性化の亢進など)を解明していくことは,ALK遺伝子異常のがんをもつ患者の治療に有用な情報をあたえるであろう.
略歴:2002年 東京大学大学院医学系研究科博士課程 修了,同年 日本ロシュ 抗癌剤研究部,同年 中外製薬 創薬研究第三部,2004年 同 創薬研究第二部を経て,2010年より同 創薬研究第二部 主席研究員.
研究テーマ:がんの分子標的薬剤の研究開発.
抱負:個々のがん患者さんにとって満足度の高い治療薬の研究開発に貢献したい.
© 2011 坂本 洋 Licensed under CC 表示 2.1 日本
(中外製薬鎌倉研究所 創薬研究第二部)
email:坂本 洋
DOI: 10.7875/first.author.2011.088
CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant.
Hiroshi Sakamoto, Toshiyuki Tsukaguchi, Sayuri Hiroshima, Tatsushi Kodama, Takamitsu Kobayashi, Takaaki A. Fukami, Nobuhiro Oikawa, Takuo Tsukuda, Nobuya Ishii, Yuko Aoki
Cancer Cell, 19, 679-690 (2011)
要 約
ALKはある種のがんにおいて染色体転座,遺伝子増幅,点変異などの遺伝子異常にともない恒常的に活性化されるチロシンキナーゼである.今回,筆者らは,これまでのキナーゼ阻害剤とは異なる骨格の化学構造をもち経口投与の可能なALK阻害剤CH5424802を同定した.このCH5424802はALKに対し強力かつ選択的なキナーゼ阻害プロファイルを示し,EML4-ALK融合遺伝子を発現する非小細胞肺がんやNPM-ALK融合遺伝子を発現する未分化大細胞型リンパ腫など,ALK遺伝子に異常をもつがん細胞に対しin vitroおよびin vivoにおいて選択的な抗腫瘍活性を示した.さらに,CH5424802はキナーゼ阻害剤に対する共通耐性機構のひとつとして知られるゲートキーパー残基での点変異に対しても有効であった.CH5424802のように耐性変異に対しても有効なALK阻害剤はALK遺伝子変異に依存性のがんをもつ患者に対し有効な治療効果をあたえる可能性が示唆された.
はじめに
最近のがんの分子標的薬剤はがんに特異的に作用し臨床においても実績をあげており,今後も新たな医薬品の開発が期待される.しかしながら,キナーゼ阻害剤においては,複数のキナーゼを同時に阻害することによる有効治療域の狭さ,あるいは,さまざまな分子機構でひき起こされる薬剤耐性の出現により,治療効果が限定的となってしまうケースがある.したがって,より強力で選択的な阻害剤,あるいは,薬剤耐性をひき起こしにくい薬剤を見い出すことが要求されている.
EML4-ALK融合遺伝子は,2007年,非小細胞肺がんにおいて新規に同定された融合がん遺伝子である1).EML4-ALK融合遺伝子の腫瘍形成能は,この融合遺伝子を導入した3T3線維芽細胞を用いた形質転換活性やこの融合遺伝子を肺胞の上皮に特異的に産生するトランスジェニックマウスにおける肺腺がんの発症により確認された1,2).また,EML4-ALK融合遺伝子には融合点の異なる複数のバリアントや3),KIF5B-ALK融合遺伝子のように融合相手の異なる融合遺伝子も同定された4)(図1).このようなALK融合遺伝子をもつ患者は非小細胞肺がん患者に約5%の頻度で認められており,EGF受容体の変異やK-ras遺伝子の変異とは相互に排他的な関係にあることが確認されている5).また,EML4-ALK融合遺伝子をもつ非小細胞肺がん患者はEGF受容体の阻害剤に対する反応に乏しいことから6),これを標的とする新たな治療法の確立が必要とされている.チロシンキナーゼであるc-METおよびALKの阻害剤であるPF-02341066(クリゾチニブ)はALK融合遺伝子をもつ非小細胞肺がん患者を対象にした臨床試験において高い奏効率を示した7).一方で,最近の報告において,クリゾチニブ処置により部分奏効したのち再発の認められた患者のがん検体を用いた遺伝子解析から,ALKにL1196MやC1156Yといった点変異がみつかった8).
今回,筆者らは,ALKに対して高い阻害選択性をもち,かつ,薬剤耐性に寄与する点変異に対しても有効な新しいタイプのALK阻害剤CH5424802を見い出した.
1.新規のALK阻害剤CH5424802とそのキナーゼ選択的な阻害
筆者らは,強力かつよりキナーゼ選択性の高いALK阻害剤を取得することに焦点をしぼり化合物の同定を試みた.キナーゼ阻害剤のハイスループットスクリーニングから新規の骨格をもつ複数のヒット化合物が得られ,それらの誘導体のなかでALKを強く阻害するリード化合物をみつけた.このリード化合物の選択性および薬物動態プロファイルを向上するため種々の化学修飾を行い,最終的に経口投与が可能で強力かつALK選択的なベンゾ[b]カルバゾール誘導体CH5424802を同定した.ALKのキナーゼ領域とCH5424802との共結晶のX線構造解析(図2,PDB ID:3AOX),および,生化学的な実験により,CH5424802がALKのATP結合部位においてATPと競合的に結合することを確認した.
in vitroでの酵素阻害活性測定系において,CH5424802のALKに対する50%阻害濃度は1.9 nMであった.また,神経芽腫細胞において活性化型の点変異として同定されているF1174LあるいはR1275Q 9) をもつALKに対しても同じ程度の阻害活性を示すことを確認した.また,CH5424802のキナーゼ選択性を確認するためさまざまなキナーゼに対する阻害活性を測定したところ,c-MET,インスリン受容体,EGF受容体などを含むALK以外の24のキナーゼに対しては弱い阻害活性を示す,あるいは,阻害活性を示さなかった.さらに,細胞を用いた実験において,CH5424802はEML4-ALK融合遺伝子を発現するNCI-H2228細胞においてALKの自己リン酸化を阻害し,それにともないSTAT3およびAKTのリン酸化の抑制も認められた.
2.ALK遺伝子変異に依存性のがんにおけるCH5424802の選択的な抗腫瘍活性
チロシンキナーゼであるEGF受容体に対する阻害剤はEGF受容体に変異をもつ患者に対し高い治療効果を示すことが確認され8),がん化に寄与するがん遺伝子産物を標的とした分子標的薬剤の有用性が期待されている.そこで,ALKおよびEGF受容体について遺伝子変異のステータスの異なる非小細胞肺がん細胞株を用い,CH5424802および比較対照としてEGF受容体の阻害剤であるゲフィチニブの抗腫瘍活性を検討した.CH5424802はEML4-ALK融合遺伝子を発現するNCI-H2228細胞において抗腫瘍活性およびアポトーシス誘導活性を示したが,ALK融合遺伝子の発現していない細胞株においてはほとんど抗腫瘍活性を示さなかった.一方,ゲフィチニブはEGF受容体に変異をもつ細胞株に対して高い抗腫瘍活性を示し,NCI-H2228細胞に対する活性は弱かった.これらの結果より,CH5424802がALK融合遺伝子をもつ非小細胞肺がんというセグメントに対し選択的に抗腫瘍活性をもたらす可能性が示された.
同様に,ALK遺伝子に異常をもつ非小細胞肺がん以外のがん細胞株におけるCH5424802の抗腫瘍活性をin vitroでの細胞増殖阻害アッセイにより評価したところ,ALK遺伝子の転座をもつ未分化大細胞型リンパ腫細胞であるKARPAS-299細胞やSR細胞,ALK遺伝子の増幅の認められる神経芽腫細胞であるNB-1細胞,ALK遺伝子に活性化型の点変異F1174LをもつKELLY細胞では,CH5424802に対する高い感受性を示した.一方,CH5424802はALK遺伝子に異常の認められない複数の細胞株では抗腫瘍効果を示さなかった.以上のことから,CH5424802は非小細胞肺がんにかぎらずALK遺伝子に異常をもつさまざまながん細胞に対し選択的に抗腫瘍活性を示すことが明らかになった.
CH5424802のin vivoにおける抗腫瘍効果について細胞株をマウスの皮下に移植したゼノグラフトモデルを用いて評価した.その結果,EML4-ALK融合遺伝子を発現する非小細胞肺がん細胞株NCI-H2228を用いたモデルにおいて用量に依存的な腫瘍増殖の抑制効果と腫瘍の退縮が認められた.この試験におけるマウスの体重減少は認められなかった.一方,ALK融合遺伝子を発現していない非小細胞肺がん細胞株A549を移植したモデルではCH5424802による抗腫瘍効果は認められなかった.NCI-H2228細胞モデルの腫瘍において標的阻害の指標となるALKのリン酸化レベルを測定したところ,CH5424802の投与によりこれが抑制されることも確認した.さらに,やや大きなサイズの腫瘍(340~350 mm3)を用いたNCI-H2228細胞のゼノグラフトモデルにおいて,3週間にわたり60 mg/kgのCH5424802を投与したのち4週間にわたり経過観察したところ,強い抗腫瘍効果は維持され腫瘍の再増殖は認められなかった.また,同様な実験をKARPAS-299細胞やNB-1細胞を用いたゼノグラフトモデルでも評価したところ,腫瘍増殖の抑制効果と腫瘍の退縮が認められた.このように,ALK遺伝子に異常をもつ複数のがん種においてCH5424802のin vivoでの高い抗腫瘍活性が確認された.
ALKのシグナル伝達系に関して,NPM-ALK融合遺伝子を発現する未分化大細胞型リンパ腫においては,その下流のタンパク質として転写因子STAT3が細胞の生存や増殖に重要な役割をはたしていることが知られている10).一方,EML4-ALK融合遺伝子を発現する非小細胞肺がんにおいてはこのような下流のタンパク質についてあまり明確には解析されていない.そこで,CH5424802を処理したのちのNCI-H2228細胞モデルの腫瘍を用いて網羅的な遺伝子解析を行ったところ,複数のSTAT3の標的遺伝子の発現低下が確認された.また,リン酸化レベルの解析において,CH5424802を投与したのちの腫瘍におけるSTAT3のリン酸化の阻害にくわえ,AKTの部分的なリン酸化の抑制も認められた.siRNAを用いた実験において,NPM-ALK融合遺伝子を発現するKARPAS-299細胞ではSTAT3のノックダウンにより十分な細胞の増殖阻害が認められたのに対し,EML4-ALK融合遺伝子を発現するNCI-H2228細胞ではSTAT3のノックダウンは細胞増殖にほとんど影響をあたえなかった.したがって,EML4-ALK融合遺伝子をもつ非小細胞肺がんにおいては,STAT3以外,もしくは,STAT3を含めた複数の下流シグナル経路が細胞増殖に必要である可能性が示唆された.
3.ALKのゲートキーパー耐性変異に対するCH5424802の効果
キナーゼ阻害剤に対する獲得耐性機構のひとつとして,キナーゼドメインにおける点変異が知られている.とりわけ,EGF受容体の点変異T790M,あるいは,BCR-ABL融合タンパク質の点変異T315Iのようなゲートキーパー残基の変異は高い頻度で起こりやすい耐性機構のひとつである.最近の報告により,クリゾチニブの臨床試験において部分奏効したのちに再発の認められた非小細胞肺がん患者のがん検体を用いた遺伝子解析から,EML4-ALK融合タンパク質の点変異L1196M(ゲートキーパー残基における変異)およびC1156Yがみつかっている8).この点変異L1196MをもつALKタンパク質に対するCH5424802の影響を調べたところ,変異をもたないALKと同じ程度の高い酵素阻害活性が認められた.
細胞における影響を試みるため,Ba/F3細胞に野生型のEML4-ALK融合遺伝子および点変異L1196MをもつEML4-ALK融合遺伝子を遺伝子導入した安定細胞株を作製した.ALKに非依存的な親細胞Ba/F3細胞と比較して,野生型あるいは点変異L1196MをもつEML4-ALK融合遺伝子を導入したBa/F3細胞においてはCH5424802に対しいずれも高い感受性を示した.一方,クリゾチニブについては,点変異L1196MをもつEML4-ALK融合遺伝子を導入したBa/F3細胞に対する感受性は親細胞と同じ程度であり,ALKの阻害による影響はほとんど認められなかった.また,この感受性に対する影響は細胞におけるALKのリン酸化レベルの抑制効果と一致していた.さらに,この抗腫瘍活性をin vivoで確認するため,これらの細胞株をマウスに移植したゼノグラフトモデルを用いて評価した.その結果,CH5424802は野生型および点変異L1196MをもつEML4-ALK融合遺伝子のいずれに依存する腫瘍に対しても腫瘍の退縮効果が認められた.
ゲートキーパー耐性変異である点変異L1196Mに対しCH5424802が阻害効果を示した理由を明らかにするため,それらの共結晶のX線構造を用いてCH5424802とALKとの結合様式を確認した.その結果,CH5424802の3位シアノ基とALKのゲートキーパー残基L1196とが効果的なCH/π相互作用をしていることが観察された.また,in silicoモデルの解析により,点変異L1196MをもつALKにおいても同様にこの効果的なCH/π相互作用が維持されていることが示唆された.一方,クリゾチニブについては,L1196残基,および,in silicoモデルにおけるM1196残基との効果的な相互作用は認められなかった.このように,共結晶構造の情報からCH5424802がALKのゲートキーパー耐性変異である点変異L1196Mに対し酵素阻害能を保持できることが考察された.
おわりに
現在,CH5424802についてはALK融合遺伝子をもつ非小細胞肺がん患者を対象とした第1相/第2相の臨床試験が進行中である.今回,見い出された新規のALK阻害剤CH5424802の特徴のひとつは,ALKに対し高いキナーゼ選択性をもつことである.低分子阻害剤のオフターゲットによる毒性は投与量を上げることのさまたげとなり,最終的に薬効を限定的にしてしまうことがある.成人の正常組織においてALKの発現はほとんど認められておらず,ALKを阻害することによる正常組織への影響は非常に小さいものと考えられるので,より選択的なALK阻害剤は広い治療域を生み出し高い効果が期待される.CH5424802のもうひとつの特徴として,ALK阻害剤に対する耐性機構のひとつであるALKの点変異に対しても有効であることがある.CH5424802は点変異としてL1196Mだけでなく,耐性変異として同様に同定された点変異C1156Yに対しても,in vitroでの酵素阻害,および,点変異C1156YをもつEML4-ALK融合タンパク質に依存した細胞の増殖阻害が確認された.CH5424802はALKに対する阻害が強力でかつ広い有効治療域を兼ね備えていることから,別の新たな点変異により阻害活性がやや落ちたとしても薬効には大きな影響のないものと考えられる.このようなことから,CH5424802はALK阻害剤に対し薬剤耐性をひき起こした患者に対しても効果があるかもしれない.今後,ALK遺伝子の点変異にかぎらず,ALK阻害剤のもつ別の耐性機構(たとえば,ほかのがん遺伝子の遺伝子異常やほかのタンパク質の活性化の亢進など)を解明していくことは,ALK遺伝子異常のがんをもつ患者の治療に有用な情報をあたえるであろう.
文 献
- Soda, M., Choi, Y. L., Enomoto, M. et al.: Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature, 448, 561-566 (2007)[PubMed]
- Soda, M., Takada, S., Takeuchi, K. et al.: A mouse model for EML4-ALK-positive lung cancer. Proc. Natl. Acad. Sci. USA, 105, 19893-19897 (2008)[PubMed]
- Takeuchi, K., Choi, Y. L., Soda, M. et al.: Multiplex reverse transcription-PCR screening for EML4-ALK fusion transcripts. Clin. Cancer Res., 14, 6618-6624 (2008)[PubMed]
- Takeuchi, K., Choi, Y. L., Togashi, Y. et al.: KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin. Cancer Res., 15, 3143-3149 (2009)[PubMed]
- Shaw, A. T., Yeap, B. Y., Mino-Kenudson, M. et al.: Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J. Clin. Oncol., 27, 4247-4253 (2009)[PubMed]
- Inamura, K., Takeuchi, K., Togashi, Y. et al.: EML4-ALK fusion is linked to histological characteristics in a subset of lung cancers. J. Thorac. Oncol., 3, 13-17 (2008)[PubMed]
- Kwak, E. L., Bang, Y., Camidge, D. R. et al.: Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Eng. J. Med., 363, 1693-1703 (2010)[PubMed]
- Choi, Y. L., Soda, M., Yamashita, Y. et al.: EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Eng. J. Med., 363, 1734-1739 (2010)[PubMed]
- Mosse, Y. P., Laudenslager, M., Longo, L. et al.: Identification of ALK as a major familial neuroblastoma predisposition gene. Nature, 455, 930-935 (2008)[PubMed]
- Chiarle, R., Simmons, W. J., Cai, H. et al.: Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat. Med., 11, 623-629 (2005)[PubMed]
著者プロフィール
略歴:2002年 東京大学大学院医学系研究科博士課程 修了,同年 日本ロシュ 抗癌剤研究部,同年 中外製薬 創薬研究第三部,2004年 同 創薬研究第二部を経て,2010年より同 創薬研究第二部 主席研究員.
研究テーマ:がんの分子標的薬剤の研究開発.
抱負:個々のがん患者さんにとって満足度の高い治療薬の研究開発に貢献したい.
© 2011 坂本 洋 Licensed under CC 表示 2.1 日本
