JNKを介した貪食機構による腫瘍原性をもつ細胞の排除
大澤志津江・井垣達吏
(神戸大学大学院医学研究科 細胞生物学分野)
email:井垣達吏
DOI: 10.7875/first.author.2011.074
Elimination of oncogenic neighbors by JNK-mediated engulfment in Drosophila.
Shizue Ohsawa, Kaoru Sugimura, Kyoko Takino, Tian Xu, Atsushi Miyawaki, Tatsushi Igaki
Developmental Cell, 20, 315-328 (2011)
多細胞生物の上皮組織に腫瘍原性の異常細胞が生じると,組織はそれを積極的に認識し排除することでその恒常性を保つと考えられる.近年のショウジョウバエを用いた遺伝学的な解析により,ショウジョウバエの上皮に生じた腫瘍原性をもつ極性の崩壊した細胞は,それをとりまく正常な細胞と“細胞競合”をひき起こし,極性の崩壊した細胞がこの競合の“敗者”となって細胞死を起こし組織から排除されることがわかってきた.今回,筆者らは,このショウジョウバエ細胞競合モデルにおいて正常な細胞が極性の崩壊した細胞の排除を促進する分子機構を解析した.その結果,正常な上皮細胞は極性の崩壊した細胞の出現に応答してアポトーシスを導かないJNKシグナルを活性化させ,このシグナルが隣接する極性の崩壊した細胞の排除を促進していることを見い出した.さらに,この正常な細胞におけるJNKの活性が細胞骨格の再編成にかかわるシグナル伝達経路PVR-ELMO/Mbc経路を介して細胞の貪食能を亢進させ,これにより隣接する極性の崩壊した細胞に対しエントーシス様の細胞死を誘導することを明らかにした.
多細胞生物は細胞間相互作用を介した細胞増殖や細胞死の調節により正常な器官発生やその恒常性の維持を実現する.この細胞間相互作用を介した調節機構が破綻すると腫瘍の形成さらにはその悪性化がひき起こされるものと考えられる.
がんのほとんどをしめる上皮由来がんは単一あるいは数個の上皮細胞が腫瘍原性の突然変異(がん遺伝子の活性化やがん抑制遺伝子の不活性化)を多段階的に蓄積することにより発生するものと考えられている1).したがって,上皮がんの発生の初期段階においては腫瘍原性をもつ異常な細胞がその周囲を正常な細胞にとりかこまれた状態で存在し,両者のあいだで正常な細胞どうしとは異なる種類の細胞間相互作用を生じているものと考えられる.興味深いことに,がん細胞をとりまく正常な細胞はしばしば抗腫瘍性の選択圧を発揮することが知られている.たとえば,リンパ球やナチュラルキラー細胞などの免疫細胞は上皮由来がんの発生を抑制する.また,がん遺伝子Rasを発現したラット胚線維芽細胞がその周囲を正常な細胞にとりかこまれると生存能が低下することや,Ras遺伝子あるいはSrc遺伝子を発現したイヌ腎上皮細胞に由来するMDCK細胞がその周囲を正常な細胞にとりかこまれると単層培養からはじきだされることが報告されている.
このような正常な細胞が腫瘍原性をもつ細胞に対して拮抗的にはたらく現象はショウジョウバエの上皮においても観察される.ショウジョウバエの成虫原基の上皮組織において腫瘍原性をもつ極性の崩壊した細胞がその周囲を正常な細胞にかこまれると,これら極性の崩壊した細胞は細胞死を起こして組織から排除される.ここで,極性の崩壊した細胞は周囲を正常な細胞にかこまれない状況においては細胞死を起こすことなくむしろ強い増殖能を発揮することから,これは隣接した細胞の性質によって細胞自体の運命が変わる(細胞死を起こす)という“細胞競合”を介した内在性のがん抑制機構であると考えられる.
この研究では,内在性のがん抑制機構を駆動する正常な細胞と腫瘍原性をもつ細胞とのあいだの細胞間コミュニケーション機構を明らかにするため,腫瘍原性をもつ細胞をとりかこむ正常な細胞の役割とその分子基盤を解析した.
上皮細胞は明確な頂端-基底軸方向の極性をもっており,この極性の崩壊はがんの発生や進行に深く関与しているものと考えられている.たとえば,さまざまな実験系において,極性の崩壊した細胞は高い増殖能を発揮する.また,頂端-基底軸方向の極性の崩壊とがんの悪性度とが正の相関を示すことも知られている.このような“極性の崩壊が増殖能を促進する”現象はショウジョウバエの上皮においても観察される.進化的に保存された頂端-基底軸方向の極性遺伝子であるscribble(scrib)遺伝子やdiscs large(dlg)遺伝子の変異体では上皮細胞が過剰に増殖して腫瘍を形成する.ところが近年,これらの極性の崩壊した細胞はその周囲を正常な細胞にかこまれると細胞死を起こすことが明らかになってきた2-6).すなわち,極性の崩壊した細胞はそれ自体が高い増殖能をもっているが,その周囲に正常な細胞が存在する状況では増殖能を発揮できなくなり細胞死により組織から排除される.このような“状況依存的”細胞死は,組織においてとなりあう細胞どうしが生存能や増殖能を競い合い,この競合の“敗者”が細胞死によって組織から排除される“細胞競合”現象と合致する7).最近,筆者らを含む複数のグループにより,この細胞競合を介した極性の崩壊した細胞の細胞死がJNKに依存的な細胞死シグナルに担われていることが明らかにされた3,4).今回,筆者らは,この現象において正常な細胞がいかにして極性の崩壊した細胞の排除を誘発するのか,その機構を正常な細胞側の役割に着目して解析を進めた.
極性の崩壊した細胞に隣接する正常な細胞のふるまいを詳細に観察した結果,JNKシグナルが極性の崩壊した細胞のみならず,それをとりかこむ正常な細胞においても活性化していることがわかった.JNKを活性化した正常な細胞は細胞死を起こしていなかったことから,正常な細胞でのJNKの活性はなんらかの非細胞死の機能をはたしているものと考えられた.JNKシグナルは細胞死のみならず細胞の増殖や分化,移動,形態変化などさまざまなプロセスを制御することが示されている.そこで,この正常な細胞側でのJNKシグナルの役割を調べるため正常な細胞でのみJNKの活性を抑制したところ,極性の崩壊した細胞の排除が抑制されることがわかった.逆に,正常な細胞においてのみJNK活性を高めると極性の崩壊した細胞の排除が亢進した.これらの結果から,正常な細胞におけるJNKの活性化は極性の崩壊した細胞の排除に促進的にはたらいていることがわかった.
正常な細胞におけるJNKシグナルの標的遺伝子を探索した結果,ショウジョウバエPDGF/VEGF受容体ホモログであるPVRをコードする遺伝子をJNKシグナルの新たなターゲットとして同定した.受容体型チロシンキナーゼであるPVRはリガンドの非存在下においてもその発現量に依存して下流シグナルを活性化しうることが知られている.正常な細胞におけるPVRの役割を遺伝学的に検証したところ,極性の崩壊した細胞をとりまく正常な細胞はJNK-PVR経路により極性の崩壊した細胞の排除を促進していることがわかった.
では,正常な細胞におけるJNK-PVRシグナルはどのようにして隣接する極性の崩壊した細胞の排除を促進するのだろうか? この疑問に答えるため極性の崩壊した細胞について細胞死の空間的なパターンを詳細に解析したところ,そのほとんどが正常な細胞クローンとの境界に存在することを見い出した.この細胞死の空間的なパターンは細胞競合に特有の現象であり,細胞競合をひき起こすことが知られているMinute変異細胞クローンやdmyc過剰発現細胞クローンを組織に誘導した際にも観察される.さらに,このような細胞競合に特有の細胞死パターンにくわえて,多くの極性の崩壊した細胞が正常な細胞クローンのなかに取り込まれるかたちで入り込んで細胞死を起こしていることに気づいた.
そこで,極性の崩壊した細胞が組織から排除される現象をリアルタイムイメージングにより追跡した結果,これらの極性の崩壊した細胞は確かにクローンから離脱し正常な細胞に取り込まれたあとばらばらになって細胞死を起こしていることがわかった.このようなクローンから離脱した極性の崩壊した細胞は貪食マーカーであるlysotrackerにより細胞の全体が標識されたことから,極性の崩壊した細胞は正常な細胞によって貪食されることにより細胞死を起こしている可能性が示唆された.同様の“貪食による細胞死誘導”は分裂速度の遅いMinute変異細胞が組織から排除される細胞競合においても観察される8).
興味深いことに,貪食の過程で細胞骨格の再編成に必須の役割をはたすELMO(Ced-12ホモログ)/Mbc(Ced-5/DOCK180ホモログ)は,ショウジョウバエの卵巣でのボーダー細胞の移動やショウジョウバエの変態のときの胸部閉鎖(thorax closure)の過程においてPVRの下流ではたらくことが知られている9,10).そこで,正常な細胞におけるELMO/Mbcの機能と役割を遺伝学的に検証した.その結果,ELMO/Mbcは正常な細胞においてJNK-PVRシグナルの下流ではたらき極性の崩壊した細胞の排除を促進していることが明らかになった.すなわち,正常な細胞は極性の崩壊した細胞の出現に応答してJNKシグナルを活性化させ,これによりPVR-ELMO/Mbc経路を介して貪食能を亢進することで,となりの極性の崩壊した細胞を生きたまま貪食して細胞死にいたらせているものと考えられた.
以上の結果から,正常な上皮組織は組織に極性を崩壊した異常な細胞が生じると異常な細胞とそのとなりの正常な細胞の両方でJNKシグナルを活性化させ,これが極性の崩壊した細胞においては細胞死シグナルとして,正常な細胞においては貪食能の亢進シグナルとして機能することで効率的な細胞排除の機構を成立させているものと考えられた(図1).
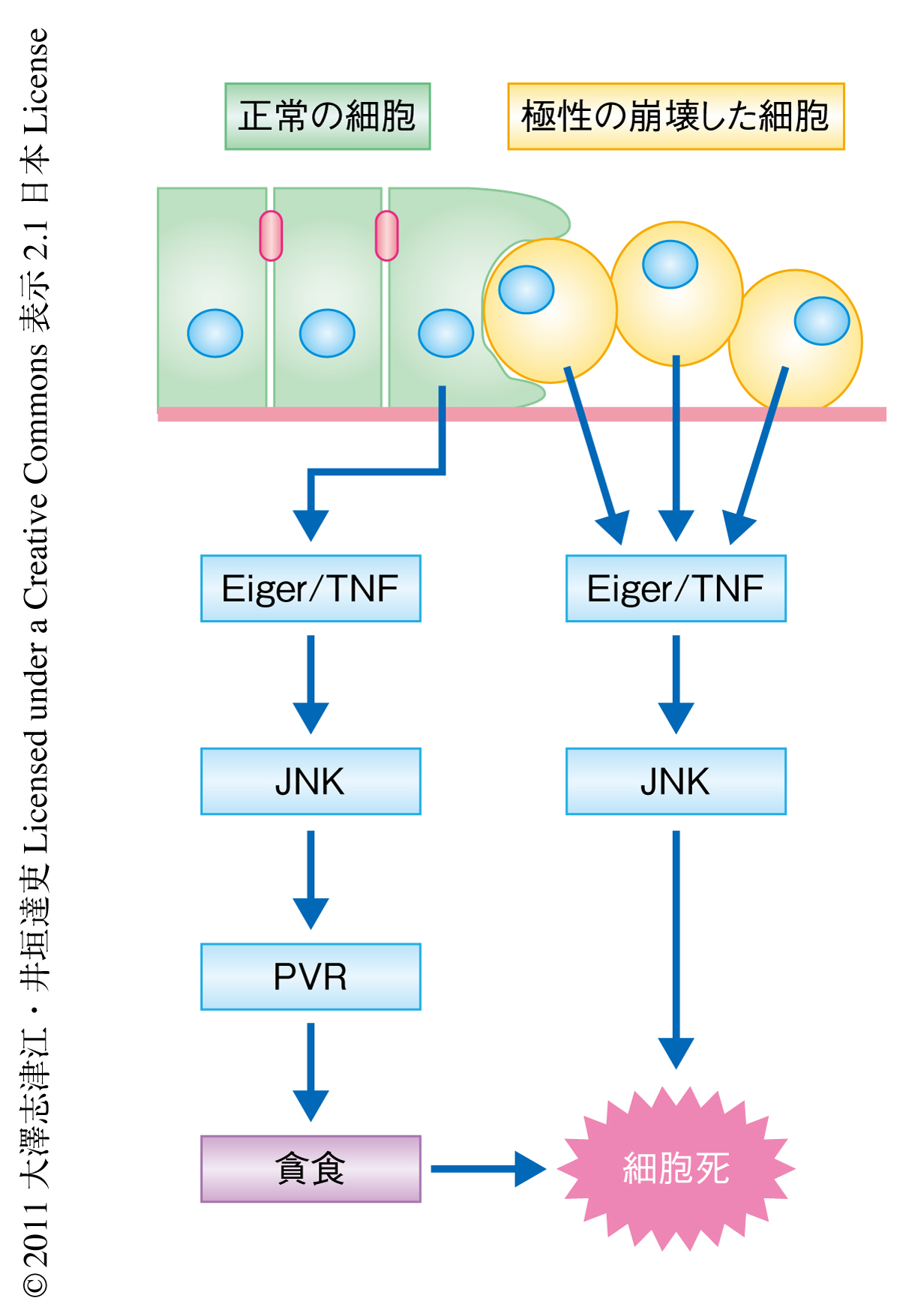
この研究により,正常組織は腫瘍原性をもつ極性の崩壊した細胞の出現に応答してJNKを介した貪食機構を活性化し,これにより細胞競合を介した“内在性のがん抑制”機構を発動することが示された.従来,貪食は組織に生じた死細胞をすみやかに組織から排除する機構であると考えられてきたが,近年,貪食が細胞死を積極的に誘導するという報告も蓄積されつつある.この研究で見い出された貪食による細胞排除の現象は,最近,哺乳類において同定された“エントーシス”に類似したものであると考えられる11).エントーシスとは細胞が同種あるいは異種の生きた細胞を飲み込む現象であり,飲み込まれた細胞はリソソーム経路により分解されることで細胞死を起こす.興味深いことに,エントーシスはヒトの転移性乳腺がんをはじめとしたさまざまながん組織で観察される.また,三次元培養したヒト乳腺がん細胞MCF7細胞はエントーシスを阻害した条件ではコロニーの形成能を上昇させることから,エントーシスががん細胞の排除現象に機能している可能性が考えられている11).この研究で同定されたショウジョウバエの正常な上皮細胞による腫瘍原性をもつ細胞の貪食にかかわるタンパク質Eiger,JNK,PVR,および,ELMO/Mbcは,いずれもヒトにおいて高度に保存されていることから,これらを介した貪食機構が進化的に保存された上皮における内在性のがん抑制機構として機能している可能性が考えられる.
略歴:2008年 東京大学大学院薬学系研究科博士後期課程 修了,同年より神戸大学大学院医学研究科 グローバルCOE研究員.
研究テーマ:細胞間コミュニケーションを介した組織の成長とその破綻による腫瘍の形成および悪性化の分子基盤の解明.
抱負:多細胞コミュニティを構成する細胞どうしが互いに影響しあい,正確な大きさやかたちの組織を形成していく分子機構を明らかにしたいと思っています.
井垣 達吏(Tatsushi Igaki)
神戸大学大学院医学研究科 特命准教授.
研究室URL:http://www.med.kobe-u.ac.jp/igalab/index.html
© 2011 大澤志津江・井垣達吏 Licensed under CC 表示 2.1 日本
(神戸大学大学院医学研究科 細胞生物学分野)
email:井垣達吏
DOI: 10.7875/first.author.2011.074
Elimination of oncogenic neighbors by JNK-mediated engulfment in Drosophila.
Shizue Ohsawa, Kaoru Sugimura, Kyoko Takino, Tian Xu, Atsushi Miyawaki, Tatsushi Igaki
Developmental Cell, 20, 315-328 (2011)
要 約
多細胞生物の上皮組織に腫瘍原性の異常細胞が生じると,組織はそれを積極的に認識し排除することでその恒常性を保つと考えられる.近年のショウジョウバエを用いた遺伝学的な解析により,ショウジョウバエの上皮に生じた腫瘍原性をもつ極性の崩壊した細胞は,それをとりまく正常な細胞と“細胞競合”をひき起こし,極性の崩壊した細胞がこの競合の“敗者”となって細胞死を起こし組織から排除されることがわかってきた.今回,筆者らは,このショウジョウバエ細胞競合モデルにおいて正常な細胞が極性の崩壊した細胞の排除を促進する分子機構を解析した.その結果,正常な上皮細胞は極性の崩壊した細胞の出現に応答してアポトーシスを導かないJNKシグナルを活性化させ,このシグナルが隣接する極性の崩壊した細胞の排除を促進していることを見い出した.さらに,この正常な細胞におけるJNKの活性が細胞骨格の再編成にかかわるシグナル伝達経路PVR-ELMO/Mbc経路を介して細胞の貪食能を亢進させ,これにより隣接する極性の崩壊した細胞に対しエントーシス様の細胞死を誘導することを明らかにした.
はじめに
多細胞生物は細胞間相互作用を介した細胞増殖や細胞死の調節により正常な器官発生やその恒常性の維持を実現する.この細胞間相互作用を介した調節機構が破綻すると腫瘍の形成さらにはその悪性化がひき起こされるものと考えられる.
がんのほとんどをしめる上皮由来がんは単一あるいは数個の上皮細胞が腫瘍原性の突然変異(がん遺伝子の活性化やがん抑制遺伝子の不活性化)を多段階的に蓄積することにより発生するものと考えられている1).したがって,上皮がんの発生の初期段階においては腫瘍原性をもつ異常な細胞がその周囲を正常な細胞にとりかこまれた状態で存在し,両者のあいだで正常な細胞どうしとは異なる種類の細胞間相互作用を生じているものと考えられる.興味深いことに,がん細胞をとりまく正常な細胞はしばしば抗腫瘍性の選択圧を発揮することが知られている.たとえば,リンパ球やナチュラルキラー細胞などの免疫細胞は上皮由来がんの発生を抑制する.また,がん遺伝子Rasを発現したラット胚線維芽細胞がその周囲を正常な細胞にとりかこまれると生存能が低下することや,Ras遺伝子あるいはSrc遺伝子を発現したイヌ腎上皮細胞に由来するMDCK細胞がその周囲を正常な細胞にとりかこまれると単層培養からはじきだされることが報告されている.
このような正常な細胞が腫瘍原性をもつ細胞に対して拮抗的にはたらく現象はショウジョウバエの上皮においても観察される.ショウジョウバエの成虫原基の上皮組織において腫瘍原性をもつ極性の崩壊した細胞がその周囲を正常な細胞にかこまれると,これら極性の崩壊した細胞は細胞死を起こして組織から排除される.ここで,極性の崩壊した細胞は周囲を正常な細胞にかこまれない状況においては細胞死を起こすことなくむしろ強い増殖能を発揮することから,これは隣接した細胞の性質によって細胞自体の運命が変わる(細胞死を起こす)という“細胞競合”を介した内在性のがん抑制機構であると考えられる.
この研究では,内在性のがん抑制機構を駆動する正常な細胞と腫瘍原性をもつ細胞とのあいだの細胞間コミュニケーション機構を明らかにするため,腫瘍原性をもつ細胞をとりかこむ正常な細胞の役割とその分子基盤を解析した.
1.正常な上皮細胞は極性の崩壊した細胞の出現に応答してアポトーシスを導かないなJNKシグナルを活性化しこれらの細胞の排除を促進する
上皮細胞は明確な頂端-基底軸方向の極性をもっており,この極性の崩壊はがんの発生や進行に深く関与しているものと考えられている.たとえば,さまざまな実験系において,極性の崩壊した細胞は高い増殖能を発揮する.また,頂端-基底軸方向の極性の崩壊とがんの悪性度とが正の相関を示すことも知られている.このような“極性の崩壊が増殖能を促進する”現象はショウジョウバエの上皮においても観察される.進化的に保存された頂端-基底軸方向の極性遺伝子であるscribble(scrib)遺伝子やdiscs large(dlg)遺伝子の変異体では上皮細胞が過剰に増殖して腫瘍を形成する.ところが近年,これらの極性の崩壊した細胞はその周囲を正常な細胞にかこまれると細胞死を起こすことが明らかになってきた2-6).すなわち,極性の崩壊した細胞はそれ自体が高い増殖能をもっているが,その周囲に正常な細胞が存在する状況では増殖能を発揮できなくなり細胞死により組織から排除される.このような“状況依存的”細胞死は,組織においてとなりあう細胞どうしが生存能や増殖能を競い合い,この競合の“敗者”が細胞死によって組織から排除される“細胞競合”現象と合致する7).最近,筆者らを含む複数のグループにより,この細胞競合を介した極性の崩壊した細胞の細胞死がJNKに依存的な細胞死シグナルに担われていることが明らかにされた3,4).今回,筆者らは,この現象において正常な細胞がいかにして極性の崩壊した細胞の排除を誘発するのか,その機構を正常な細胞側の役割に着目して解析を進めた.
極性の崩壊した細胞に隣接する正常な細胞のふるまいを詳細に観察した結果,JNKシグナルが極性の崩壊した細胞のみならず,それをとりかこむ正常な細胞においても活性化していることがわかった.JNKを活性化した正常な細胞は細胞死を起こしていなかったことから,正常な細胞でのJNKの活性はなんらかの非細胞死の機能をはたしているものと考えられた.JNKシグナルは細胞死のみならず細胞の増殖や分化,移動,形態変化などさまざまなプロセスを制御することが示されている.そこで,この正常な細胞側でのJNKシグナルの役割を調べるため正常な細胞でのみJNKの活性を抑制したところ,極性の崩壊した細胞の排除が抑制されることがわかった.逆に,正常な細胞においてのみJNK活性を高めると極性の崩壊した細胞の排除が亢進した.これらの結果から,正常な細胞におけるJNKの活性化は極性の崩壊した細胞の排除に促進的にはたらいていることがわかった.
2.正常な細胞はJNKの下流でPVRを誘導することにより極性の崩壊した細胞の排除を促進する
正常な細胞におけるJNKシグナルの標的遺伝子を探索した結果,ショウジョウバエPDGF/VEGF受容体ホモログであるPVRをコードする遺伝子をJNKシグナルの新たなターゲットとして同定した.受容体型チロシンキナーゼであるPVRはリガンドの非存在下においてもその発現量に依存して下流シグナルを活性化しうることが知られている.正常な細胞におけるPVRの役割を遺伝学的に検証したところ,極性の崩壊した細胞をとりまく正常な細胞はJNK-PVR経路により極性の崩壊した細胞の排除を促進していることがわかった.
3.正常な細胞のJNK-PVRシグナルはELMO/Mbcを介した貪食機構を活性化することで極性の崩壊した細胞の排除を促進する
では,正常な細胞におけるJNK-PVRシグナルはどのようにして隣接する極性の崩壊した細胞の排除を促進するのだろうか? この疑問に答えるため極性の崩壊した細胞について細胞死の空間的なパターンを詳細に解析したところ,そのほとんどが正常な細胞クローンとの境界に存在することを見い出した.この細胞死の空間的なパターンは細胞競合に特有の現象であり,細胞競合をひき起こすことが知られているMinute変異細胞クローンやdmyc過剰発現細胞クローンを組織に誘導した際にも観察される.さらに,このような細胞競合に特有の細胞死パターンにくわえて,多くの極性の崩壊した細胞が正常な細胞クローンのなかに取り込まれるかたちで入り込んで細胞死を起こしていることに気づいた.
そこで,極性の崩壊した細胞が組織から排除される現象をリアルタイムイメージングにより追跡した結果,これらの極性の崩壊した細胞は確かにクローンから離脱し正常な細胞に取り込まれたあとばらばらになって細胞死を起こしていることがわかった.このようなクローンから離脱した極性の崩壊した細胞は貪食マーカーであるlysotrackerにより細胞の全体が標識されたことから,極性の崩壊した細胞は正常な細胞によって貪食されることにより細胞死を起こしている可能性が示唆された.同様の“貪食による細胞死誘導”は分裂速度の遅いMinute変異細胞が組織から排除される細胞競合においても観察される8).
興味深いことに,貪食の過程で細胞骨格の再編成に必須の役割をはたすELMO(Ced-12ホモログ)/Mbc(Ced-5/DOCK180ホモログ)は,ショウジョウバエの卵巣でのボーダー細胞の移動やショウジョウバエの変態のときの胸部閉鎖(thorax closure)の過程においてPVRの下流ではたらくことが知られている9,10).そこで,正常な細胞におけるELMO/Mbcの機能と役割を遺伝学的に検証した.その結果,ELMO/Mbcは正常な細胞においてJNK-PVRシグナルの下流ではたらき極性の崩壊した細胞の排除を促進していることが明らかになった.すなわち,正常な細胞は極性の崩壊した細胞の出現に応答してJNKシグナルを活性化させ,これによりPVR-ELMO/Mbc経路を介して貪食能を亢進することで,となりの極性の崩壊した細胞を生きたまま貪食して細胞死にいたらせているものと考えられた.
以上の結果から,正常な上皮組織は組織に極性を崩壊した異常な細胞が生じると異常な細胞とそのとなりの正常な細胞の両方でJNKシグナルを活性化させ,これが極性の崩壊した細胞においては細胞死シグナルとして,正常な細胞においては貪食能の亢進シグナルとして機能することで効率的な細胞排除の機構を成立させているものと考えられた(図1).
おわりに
この研究により,正常組織は腫瘍原性をもつ極性の崩壊した細胞の出現に応答してJNKを介した貪食機構を活性化し,これにより細胞競合を介した“内在性のがん抑制”機構を発動することが示された.従来,貪食は組織に生じた死細胞をすみやかに組織から排除する機構であると考えられてきたが,近年,貪食が細胞死を積極的に誘導するという報告も蓄積されつつある.この研究で見い出された貪食による細胞排除の現象は,最近,哺乳類において同定された“エントーシス”に類似したものであると考えられる11).エントーシスとは細胞が同種あるいは異種の生きた細胞を飲み込む現象であり,飲み込まれた細胞はリソソーム経路により分解されることで細胞死を起こす.興味深いことに,エントーシスはヒトの転移性乳腺がんをはじめとしたさまざまながん組織で観察される.また,三次元培養したヒト乳腺がん細胞MCF7細胞はエントーシスを阻害した条件ではコロニーの形成能を上昇させることから,エントーシスががん細胞の排除現象に機能している可能性が考えられている11).この研究で同定されたショウジョウバエの正常な上皮細胞による腫瘍原性をもつ細胞の貪食にかかわるタンパク質Eiger,JNK,PVR,および,ELMO/Mbcは,いずれもヒトにおいて高度に保存されていることから,これらを介した貪食機構が進化的に保存された上皮における内在性のがん抑制機構として機能している可能性が考えられる.
文 献
- Hanahan, D. & Weinberg, R. A.: Hallmarks of cancer: the next generation. Cell, 144, 646-674 (2011)[PubMed]
- Agrawal, N., Kango, M., Mishra, A. et al.: Neoplastic transformation and aberrant cell-cell interactions in genetic mosaics of lethal(2)giant larvae (lgl), a tumor suppressor gene of Drosophila. Dev. Biol., 172, 218-229 (1995)[PubMed]
- Brumby, A. M. & Richardson, H. E.: scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J., 22, 5769-5779 (2003)[PubMed]
- Igaki, T., Pastor-Pareja, J. C., Aonuma, H. et al.: Intrinsic tumor suppression and epithelial maintenance by endocytic activation of Eiger/TNF signaling in Drosophila. Dev. Cell, 16, 458-465 (2009)[PubMed]
- Pagliarini, R. A. & Xu, T.: A genetic screen in Drosophila for metastatic behavior. Science, 302, 1227-1231 (2003)[PubMed]
- Woods, D. F. & Bryant, P. J.: The discs-large tumor suppressor gene of Drosophila encodes a guanylate kinase homolog localized at septate junctions. Cell, 66, 451-464 (1991)[PubMed]
- Morata, G. & Ripoll, P.: Minutes: mutants of drosophila autonomously affecting cell division rate. Dev. Biol., 42, 211-221 (1975)[PubMed]
- Li, W. & Baker, N. E.: Engulfment is required for cell competition. Cell, 129, 1215-1225 (2007)[PubMed]
- Bianco, A., Poukkula, M., Cliffe, A. et al.: Two distinct modes of guidance signalling during collective migration of border cells. Nature, 448, 362-365 (2007)[PubMed]
- Ishimaru, S., Ueda, R., Hinohara, Y. et al.: PVR plays a critical role via JNK activation in thorax closure during Drosophila metamorphosis. EMBO J., 23, 3984-3994 (2004)[PubMed]
- Overholtzer, M., Mailleux, A. A., Mouneimne, G. et al.: A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell, 131, 966-979 (2007)[PubMed]
著者プロフィール
略歴:2008年 東京大学大学院薬学系研究科博士後期課程 修了,同年より神戸大学大学院医学研究科 グローバルCOE研究員.
研究テーマ:細胞間コミュニケーションを介した組織の成長とその破綻による腫瘍の形成および悪性化の分子基盤の解明.
抱負:多細胞コミュニティを構成する細胞どうしが互いに影響しあい,正確な大きさやかたちの組織を形成していく分子機構を明らかにしたいと思っています.
井垣 達吏(Tatsushi Igaki)
神戸大学大学院医学研究科 特命准教授.
研究室URL:http://www.med.kobe-u.ac.jp/igalab/index.html
© 2011 大澤志津江・井垣達吏 Licensed under CC 表示 2.1 日本
