アディポネクチンはマクロファージに由来するインターロイキン6に依存的に肝臓においてIRS2を増加させインスリン感受性を亢進させる
粟澤元晴・植木浩二郎・門脇 孝
(東京大学大学院医学系研究科 代謝・栄養病態学)
email:粟澤元晴
DOI: 10.7875/first.author.2011.072
Adiponectin enhances insulin sensitivity by increasing hepatic IRS-2 expression via a macrophage-derived IL-6-dependent pathway.
Motoharu Awazawa, Kohjiro Ueki, Kazunori Inabe, Toshimasa Yamauchi, Naoto Kubota, Kazuma Kaneko, Masatoshi Kobayashi, Aya Iwane, Takayoshi Sasako, Yukiko Okazaki, Mitsuru Ohsugi, Iseki Takamoto, Satoshi Yamashita, Hiroshi Asahara, Shizuo Akira, Masato Kasuga, Takashi Kadowaki
Cell Metabolism, 13, 401-412 (2011)
アディポネクチンはインスリン感受性ホルモンとして知られているが,これまで,その作用はAMPキナーゼとPPARαの活性化によりおもに説明されており,アディポネクチンによるインスリンシグナル伝達タンパク質の制御についてはほとんど報告されていない.筆者らは,アディポネクチンが肝臓においてIRS2の発現を上昇させインスリン感受性を亢進させることをはじめて明らかにした.興味深いことに,このIRS2の発現上昇は既知のアディポネクチン受容体であるAdipoR1およびAdipoR2を介さず,未知のアディポネクチン受容体によるインターロイキン6の一過性の誘導により担われていた.
肝臓や筋肉といった標的臓器におけるインスリン作用の不足は“インスリン抵抗性”とよばれ,肥満やメタボリック症候群,2型糖尿病の発症の重要な背景である.インスリンシグナル伝達系における主要なタンパク質のひとつであるインスリン受容体基質(insulin receptor substrate:IRS)は,肥満にともないさまざまな修飾や発現抑制をうけインスリン抵抗性の惹起に関与する1,2).とくに肝臓においては,インスリン受容体基質のうちIRS2の発現が選択的に低下し,肥満モデルマウスにおけるインスリン抵抗性に関与していることが知られている3).
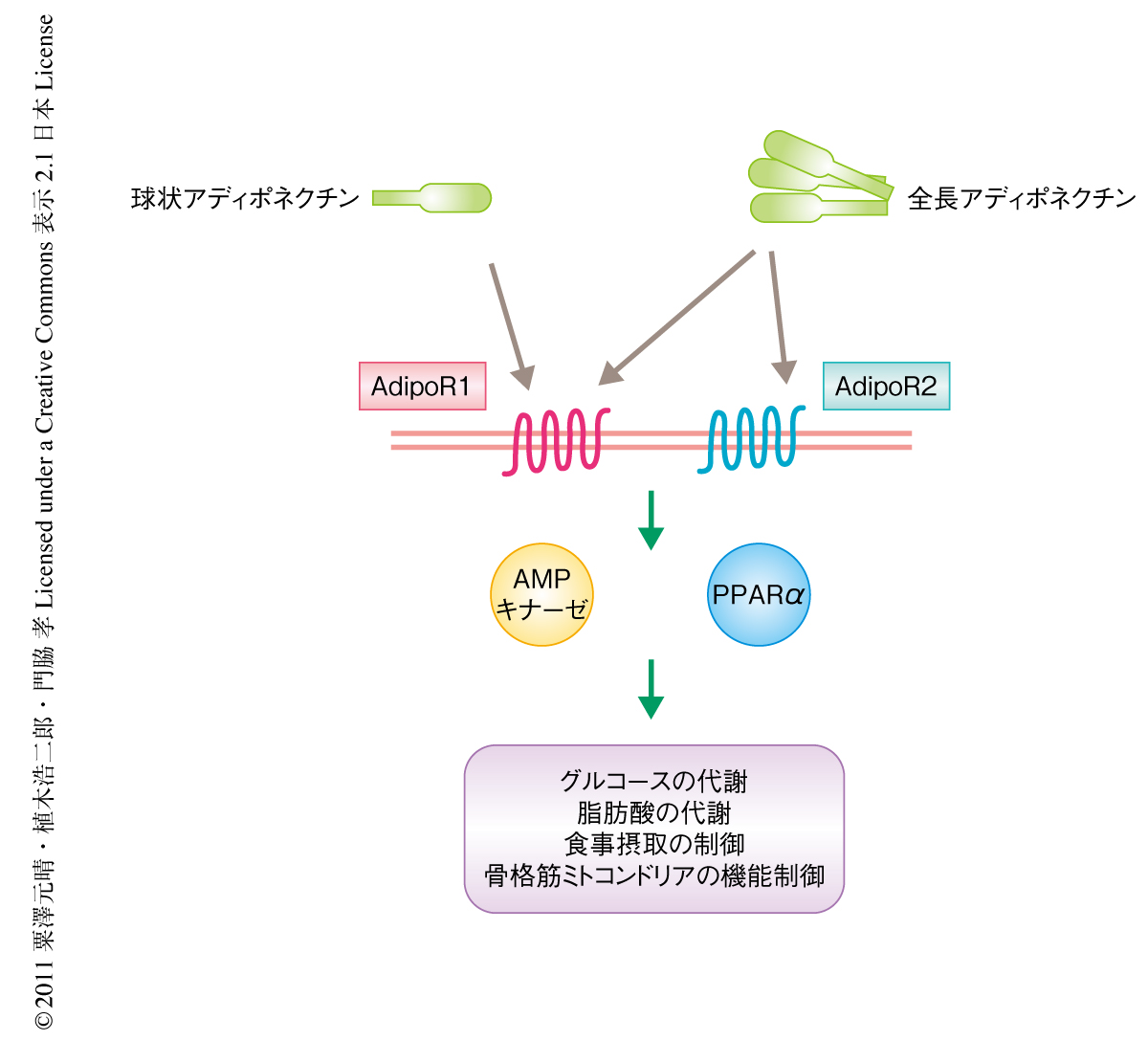
脂肪組織は生体のおかれた環境に応じてさまざまな液性因子を分泌しており,これら液性因子を介し全身の臓器に作用することで生体における恒常性の維持に関与している4).これらの液性因子はアディポカインと総称されるが,そのひとつであるアディポネクチンはインスリン感受性ホルモンとして抗糖尿病作用をもつことが広く知られており,これまで,その作用機序についてさまざまな知見が集積されてきた5).その作用の多くはAdipoR1およびAdipoR2とよばれる2つのアディポネクチン受容体を介したAMPキナーゼおよびPPARαの2つの鍵タンパク質の活性化により説明されてきたが(図1),アディポネクチンがインスリンシグナル伝達タンパク質を直接に制御するかどうかについてはこれまでほとんど知見がなかった.
筆者らは,この研究において,アディポネクチンによるインスリンシグナル伝達タンパク質の制御機構を明らかにすることをめざして以下の実験を進めた.
アディポネクチンノックアウトマウスの肝臓においてインスリンシグナル伝達タンパク質を解析したところIRS2の発現が選択的に低下していることが見い出された.一方,アディポネクチンノックアウトマウスあるいは肥満モデルマウスの腹腔内にアディポネクチンを投与すると肝臓におけるIRS2の発現が選択的に上昇した.mRNAレベルではアディポネクチンの腹腔内への投与ののち2時間で一過性にIRS2の鋭い発現上昇が認められた.このIRS2の増加はインスリン刺激によるPI3キナーゼ活性およびAktリン酸化の改善,糖新生関連遺伝子の発現低下,ピルビン酸負荷試験における血糖値の低下をともなっており,アディポネクチンによるIRS2の発現上昇が実際にインスリン作用の改善をもたらすことが示唆された.
当初は,これらIRS2の発現上昇を介したインスリン感受性の亢進は肝臓におけるアディポネクチンによる直接の作用であるものと推測した.しかしながら,肝臓において既知のアディポネクチン受容体であるAdipoR1およびAdipoR2の発現を抑制してもアディポネクチンによるIRS2の発現上昇は減弱しなかった.また,in vitroにおいて肝細胞系培養細胞をアディポネクチンにより刺激してもIRS2の発現に変化はみられなかった.これらの結果から,これまで知られていない未知の作用機構の存在が予測された.
そこで,アディポネクチンの投与ののちのIRS2の発現上昇にさきだち肝細胞においてどのような変化が起こっているのか,複数のシグナル伝達タンパク質についてそのリン酸化を調べたところ,アディポネクチンの投与ののち2時間でSTAT3が強くリン酸化されていること,また,このSTAT3のリン酸化に一致して血中のインターロイキン6の濃度が一過性に大きく上昇していることが見い出された.その強度また時間経過がIRS2の発現上昇とよく一致していたことから,アディポネクチンによりなんらかの機序でインターロイキン6が誘導され,これが肝臓においてSTAT3をリン酸化しIRS2の発現を亢進させたのではないかとの仮説をたてた.この仮説はin vitroにおいて培養細胞をアディポネクチンにより刺激したとき,IRS2と同様にSTAT3のリン酸化の亢進もみられなかったこととも矛盾しない.
しかし,一般的にインターロイキン6はインスリン抵抗性を惹起する“悪玉”ホルモンとして知られている.この時点では,アディポネクチンがインターロイキン6を誘導し,さらに,このインターロイキン6がIRS2の発現上昇を介してインスリン感受性を亢進させるという仮説は既存の概念に反するものであった.
この仮説を検証すべく,インターロイキン6中和抗体およびインターロイキン6ノックアウトマウスを用いてアディポネクチンによるIRS2の発現上昇への影響を調べた.驚くべきことに,インターロイキン6中和抗体で前処理を行うことでインターロイキン6の作用をさまたげると,アディポネクチンによる肝臓におけるSTAT3のリン酸化およびIRS2の発現上昇は完全に消失した.そして,同様の結果がインターロイキン6ノックアウトマウスを用いた実験でも追試された.一方で,野生型マウスにおいてインターロイキン6の腹腔内への投与ののち1時間でmRNAレベルでのIRS2の一過性の発現上昇が確認された.さらに,肝細胞に特異的なSTAT3ノックアウトマウスにおいては,アディポネクチンの投与によるインターロイキン6の誘導は同様に認められたがIRS2の発現上昇は完全に消失していた.これらの結果から,アディポネクチンによる肝臓におけるIRS2の発現上昇にはインターロイキン6の誘導とそれによる肝臓でのSTAT3のリン酸化が必須であると結論づけた.
これまで,IRS2の発現にSTAT3がかかわっているという報告はない.さきの結論を裏づけるため,STAT3がIRS2の発現上昇にかかわるかどうかさらに検証を進めた.ルシフェラーゼアッセイによるプロモーター解析を行ったところ,肝細胞系培養細胞においてSTAT3の過剰発現がIrs2遺伝子のプロモーター活性を上昇させることが確認された.また,アディポネクチンを投与したマウスの肝臓を用いてクロマチン免疫沈降を行ったところ,STAT3がアディポネクチンに依存的にIrs2遺伝子のプロモーター領域に結合することが示された.プロモーター解析の結果とあわせて,STAT3のIrs2遺伝子のプロモーター領域への結合部位は転写開始点の上流550塩基対から下流850塩基対までという転写開始点の近傍にあることが推定された.
アディポネクチンの投与によりインターロイキン6の血中濃度の上昇がみられたが,このインターロイキン6はどこに由来するものであろうか? このことを調べるため以下の複数の実験を行った.肥満モデルマウスにアディポネクチンを投与したのち白色脂肪組織を分離し脂肪細胞成分と間葉系細胞成分とに分画したところ,インターロイキン6の発現はおもに間葉系成分において認められた.またこのとき,免疫組織染色によりインターロイキン6の発現部位とマクロファージに特異的なタンパク質F4/80の発現部位とは完全に一致した.培養細胞においても,マクロファージ系培養細胞ではアディポネクチンにより強いインターロイキン6の誘導がみられたが,脂肪細胞系培養細胞ではインターロイキン6は誘導されなかった.さらに,インターロイキン6ノックアウトマウスに野生型マウスの骨髄を移植すると,アディポネクチンの投与による血中のインターロイキン6の濃度上昇と肝臓におけるIRS2の発現上昇が回復した.以上より,アディポネクチンは少なくともマクロファージにおいてインターロイキン6を誘導し,このインターロイキン6の発現上昇が肝臓におけるIRS2の発現を上昇させるものと結論づけた.ただし,マクロファージ以外の臓器におけるインターロイキン6の誘導を除外することは困難であり,この点については今後の検討課題とされた.
興味深いことに,既知のアディポネクチン受容体であるAdipoR1およびAdipoR2のノックアウトマウスにおいてもアディポネクチンによるインターロイキン6の発現誘導は同様に認められたことから,マクロファージにはインターロイキン6の誘導を担う未知のアディポネクチン受容体の存在することが推定された.
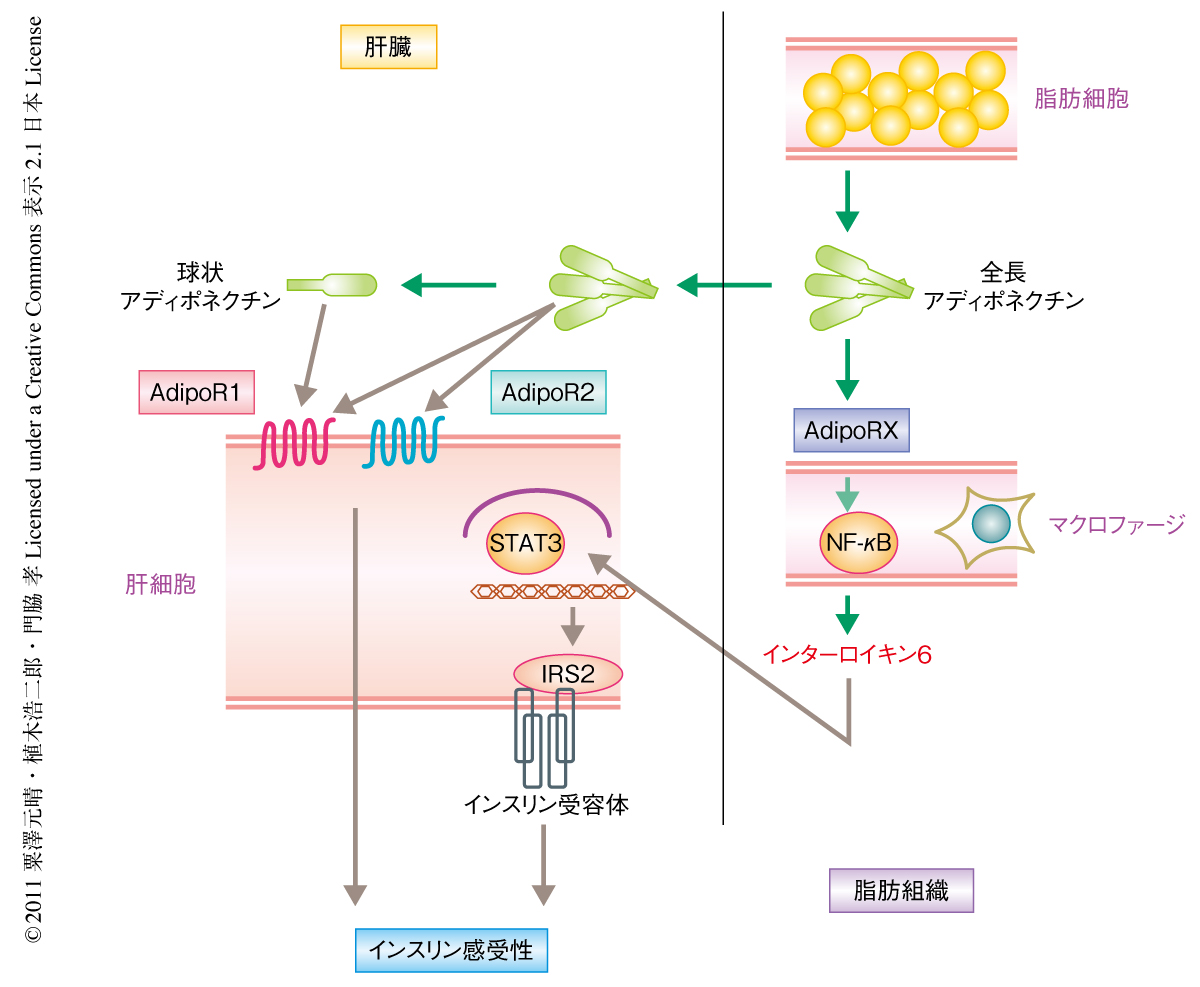
この研究により,アディポネクチンによるマクロファージからのインターロイキン6の誘導,インターロイキン6によるSTAT3を介した肝臓でのIRS2の発現上昇,という2つの新たな経路が明らかになった(図2).
肥満からインスリン抵抗性さらに糖尿病が惹起される重要な分子機構のひとつが脂肪組織における慢性炎症にあることはこの10年で広く認知されるにいたった.一般的に抗糖尿病作用をもつ“善玉”アディポカインとして知られるアディポネクチンもまた,抗炎症作用を示すことが複数の論文において報告されている6,7).ところが,一部の研究者はこれまで一定の条件下ではアディポネクチンが炎症を惹起しうることを報告しており8,9),これら矛盾する結果をどのように説明すればよいかは謎であった.この研究では未知のアディポネクチン受容体の存在が示唆され,この受容体が炎症性細胞であるマクロファージにおいてNF-κBを介してインターロイキン6の発現誘導にかかわることが明らかになった.もし,この未知の受容体を含めたアディポネクチンの複数の受容体が環境あるいは細胞に依存して異なる発現パターンをとり異なる(あるいは,逆の)作用を示すものと仮定すると,これまでの相反する結果を矛盾なく説明できるであろう.
そして,インターロイキン6の作用についてもまたいくつかの相反する報告がなされてきた.すなわち,肥満あるいはインスリン抵抗性の状態においてはインターロイキン6の血中濃度が慢性的に上昇しており,このことはインスリン抵抗性の原因のひとつとなる10).一方で,一部の研究者によりインターロイキン6の投与がインスリン感受性を亢進させる場合があると報告されていたが11),その機序は不明であり,それ以上に,肥満あるいはインスリン抵抗性の状態においてインターロイキン6の上昇していることと矛盾すると反論されてきた.この研究では,インターロイキン6の一過性の上昇がSTAT3の活性化を介して肝臓におけるIRS2の発現を上昇させ,これがインスリン感受性を亢進させることが示された.筆者らは,インターロイキン6の一過性の上昇がSTAT3を活性化することができるならば,ひき続いて肝臓におけるIRS2の発現上昇とインスリン感受性の亢進作用を惹起しうる一方で,慢性的にインターロイキン6の上昇している環境においては,おそらくはSOCS3などの負のフィードバックがはたらいてSTAT3の活性化は起こらず,むしろ,このSOCS3などがインスリンの作用を阻害しているのではないか,と提唱している.実際に,運動などにより筋肉から一過性にインターロイキン6の分泌されることが知られており12),今回の報告が,インターロイキン6の生理学的な役割についてのより踏み込んだ研究および理解につながる一助となることが期待される.
略歴:2007年 東京大学大学院医学系研究科 修了,2008年 国立健康・栄養研究所 特任研究員を経て,2009年より東京大学大学院医学系研究科 助教.
研究テーマ:アディポカインの生理学,膵β細胞における小胞体ストレス応答系.
関心事:ヒトの摂食行動を分子生物学的にどこまで説明できるか.
植木 浩二郎(Kohjiro Ueki)
東京大学大学院医学系研究科 准教授.
門脇 孝(Takashi Kadowaki)
東京大学大学院医学系研究科 教授.
© 2011 粟澤元晴・植木浩二郎・門脇 孝 Licensed under CC 表示 2.1 日本
(東京大学大学院医学系研究科 代謝・栄養病態学)
email:粟澤元晴
DOI: 10.7875/first.author.2011.072
Adiponectin enhances insulin sensitivity by increasing hepatic IRS-2 expression via a macrophage-derived IL-6-dependent pathway.
Motoharu Awazawa, Kohjiro Ueki, Kazunori Inabe, Toshimasa Yamauchi, Naoto Kubota, Kazuma Kaneko, Masatoshi Kobayashi, Aya Iwane, Takayoshi Sasako, Yukiko Okazaki, Mitsuru Ohsugi, Iseki Takamoto, Satoshi Yamashita, Hiroshi Asahara, Shizuo Akira, Masato Kasuga, Takashi Kadowaki
Cell Metabolism, 13, 401-412 (2011)
要 約
アディポネクチンはインスリン感受性ホルモンとして知られているが,これまで,その作用はAMPキナーゼとPPARαの活性化によりおもに説明されており,アディポネクチンによるインスリンシグナル伝達タンパク質の制御についてはほとんど報告されていない.筆者らは,アディポネクチンが肝臓においてIRS2の発現を上昇させインスリン感受性を亢進させることをはじめて明らかにした.興味深いことに,このIRS2の発現上昇は既知のアディポネクチン受容体であるAdipoR1およびAdipoR2を介さず,未知のアディポネクチン受容体によるインターロイキン6の一過性の誘導により担われていた.
はじめに
肝臓や筋肉といった標的臓器におけるインスリン作用の不足は“インスリン抵抗性”とよばれ,肥満やメタボリック症候群,2型糖尿病の発症の重要な背景である.インスリンシグナル伝達系における主要なタンパク質のひとつであるインスリン受容体基質(insulin receptor substrate:IRS)は,肥満にともないさまざまな修飾や発現抑制をうけインスリン抵抗性の惹起に関与する1,2).とくに肝臓においては,インスリン受容体基質のうちIRS2の発現が選択的に低下し,肥満モデルマウスにおけるインスリン抵抗性に関与していることが知られている3).
脂肪組織は生体のおかれた環境に応じてさまざまな液性因子を分泌しており,これら液性因子を介し全身の臓器に作用することで生体における恒常性の維持に関与している4).これらの液性因子はアディポカインと総称されるが,そのひとつであるアディポネクチンはインスリン感受性ホルモンとして抗糖尿病作用をもつことが広く知られており,これまで,その作用機序についてさまざまな知見が集積されてきた5).その作用の多くはAdipoR1およびAdipoR2とよばれる2つのアディポネクチン受容体を介したAMPキナーゼおよびPPARαの2つの鍵タンパク質の活性化により説明されてきたが(図1),アディポネクチンがインスリンシグナル伝達タンパク質を直接に制御するかどうかについてはこれまでほとんど知見がなかった.
筆者らは,この研究において,アディポネクチンによるインスリンシグナル伝達タンパク質の制御機構を明らかにすることをめざして以下の実験を進めた.
1.アディポネクチンは肥満モデルマウスにおいて肝臓のIRS2を増加させインスリン感受性を亢進させる
アディポネクチンノックアウトマウスの肝臓においてインスリンシグナル伝達タンパク質を解析したところIRS2の発現が選択的に低下していることが見い出された.一方,アディポネクチンノックアウトマウスあるいは肥満モデルマウスの腹腔内にアディポネクチンを投与すると肝臓におけるIRS2の発現が選択的に上昇した.mRNAレベルではアディポネクチンの腹腔内への投与ののち2時間で一過性にIRS2の鋭い発現上昇が認められた.このIRS2の増加はインスリン刺激によるPI3キナーゼ活性およびAktリン酸化の改善,糖新生関連遺伝子の発現低下,ピルビン酸負荷試験における血糖値の低下をともなっており,アディポネクチンによるIRS2の発現上昇が実際にインスリン作用の改善をもたらすことが示唆された.
2.アディポネクチンの腹腔内投与により血中のインターロイキン6の濃度が一過性に上昇し肝臓においてSTAT3のリン酸化が亢進する
当初は,これらIRS2の発現上昇を介したインスリン感受性の亢進は肝臓におけるアディポネクチンによる直接の作用であるものと推測した.しかしながら,肝臓において既知のアディポネクチン受容体であるAdipoR1およびAdipoR2の発現を抑制してもアディポネクチンによるIRS2の発現上昇は減弱しなかった.また,in vitroにおいて肝細胞系培養細胞をアディポネクチンにより刺激してもIRS2の発現に変化はみられなかった.これらの結果から,これまで知られていない未知の作用機構の存在が予測された.
そこで,アディポネクチンの投与ののちのIRS2の発現上昇にさきだち肝細胞においてどのような変化が起こっているのか,複数のシグナル伝達タンパク質についてそのリン酸化を調べたところ,アディポネクチンの投与ののち2時間でSTAT3が強くリン酸化されていること,また,このSTAT3のリン酸化に一致して血中のインターロイキン6の濃度が一過性に大きく上昇していることが見い出された.その強度また時間経過がIRS2の発現上昇とよく一致していたことから,アディポネクチンによりなんらかの機序でインターロイキン6が誘導され,これが肝臓においてSTAT3をリン酸化しIRS2の発現を亢進させたのではないかとの仮説をたてた.この仮説はin vitroにおいて培養細胞をアディポネクチンにより刺激したとき,IRS2と同様にSTAT3のリン酸化の亢進もみられなかったこととも矛盾しない.
しかし,一般的にインターロイキン6はインスリン抵抗性を惹起する“悪玉”ホルモンとして知られている.この時点では,アディポネクチンがインターロイキン6を誘導し,さらに,このインターロイキン6がIRS2の発現上昇を介してインスリン感受性を亢進させるという仮説は既存の概念に反するものであった.
3.アディポネクチンによる肝臓でのIRS2の発現上昇にはインターロイキン6の誘導と肝臓でのSTAT3のリン酸化が必須である
この仮説を検証すべく,インターロイキン6中和抗体およびインターロイキン6ノックアウトマウスを用いてアディポネクチンによるIRS2の発現上昇への影響を調べた.驚くべきことに,インターロイキン6中和抗体で前処理を行うことでインターロイキン6の作用をさまたげると,アディポネクチンによる肝臓におけるSTAT3のリン酸化およびIRS2の発現上昇は完全に消失した.そして,同様の結果がインターロイキン6ノックアウトマウスを用いた実験でも追試された.一方で,野生型マウスにおいてインターロイキン6の腹腔内への投与ののち1時間でmRNAレベルでのIRS2の一過性の発現上昇が確認された.さらに,肝細胞に特異的なSTAT3ノックアウトマウスにおいては,アディポネクチンの投与によるインターロイキン6の誘導は同様に認められたがIRS2の発現上昇は完全に消失していた.これらの結果から,アディポネクチンによる肝臓におけるIRS2の発現上昇にはインターロイキン6の誘導とそれによる肝臓でのSTAT3のリン酸化が必須であると結論づけた.
4.アディポネクチンの刺激によりSTAT3がIrs2遺伝子のプロモーター領域へ結合する
これまで,IRS2の発現にSTAT3がかかわっているという報告はない.さきの結論を裏づけるため,STAT3がIRS2の発現上昇にかかわるかどうかさらに検証を進めた.ルシフェラーゼアッセイによるプロモーター解析を行ったところ,肝細胞系培養細胞においてSTAT3の過剰発現がIrs2遺伝子のプロモーター活性を上昇させることが確認された.また,アディポネクチンを投与したマウスの肝臓を用いてクロマチン免疫沈降を行ったところ,STAT3がアディポネクチンに依存的にIrs2遺伝子のプロモーター領域に結合することが示された.プロモーター解析の結果とあわせて,STAT3のIrs2遺伝子のプロモーター領域への結合部位は転写開始点の上流550塩基対から下流850塩基対までという転写開始点の近傍にあることが推定された.
5.アディポネクチンは未知の受容体を介してマクロファージからインターロイキン6を誘導する
アディポネクチンの投与によりインターロイキン6の血中濃度の上昇がみられたが,このインターロイキン6はどこに由来するものであろうか? このことを調べるため以下の複数の実験を行った.肥満モデルマウスにアディポネクチンを投与したのち白色脂肪組織を分離し脂肪細胞成分と間葉系細胞成分とに分画したところ,インターロイキン6の発現はおもに間葉系成分において認められた.またこのとき,免疫組織染色によりインターロイキン6の発現部位とマクロファージに特異的なタンパク質F4/80の発現部位とは完全に一致した.培養細胞においても,マクロファージ系培養細胞ではアディポネクチンにより強いインターロイキン6の誘導がみられたが,脂肪細胞系培養細胞ではインターロイキン6は誘導されなかった.さらに,インターロイキン6ノックアウトマウスに野生型マウスの骨髄を移植すると,アディポネクチンの投与による血中のインターロイキン6の濃度上昇と肝臓におけるIRS2の発現上昇が回復した.以上より,アディポネクチンは少なくともマクロファージにおいてインターロイキン6を誘導し,このインターロイキン6の発現上昇が肝臓におけるIRS2の発現を上昇させるものと結論づけた.ただし,マクロファージ以外の臓器におけるインターロイキン6の誘導を除外することは困難であり,この点については今後の検討課題とされた.
興味深いことに,既知のアディポネクチン受容体であるAdipoR1およびAdipoR2のノックアウトマウスにおいてもアディポネクチンによるインターロイキン6の発現誘導は同様に認められたことから,マクロファージにはインターロイキン6の誘導を担う未知のアディポネクチン受容体の存在することが推定された.
おわりに
この研究により,アディポネクチンによるマクロファージからのインターロイキン6の誘導,インターロイキン6によるSTAT3を介した肝臓でのIRS2の発現上昇,という2つの新たな経路が明らかになった(図2).
肥満からインスリン抵抗性さらに糖尿病が惹起される重要な分子機構のひとつが脂肪組織における慢性炎症にあることはこの10年で広く認知されるにいたった.一般的に抗糖尿病作用をもつ“善玉”アディポカインとして知られるアディポネクチンもまた,抗炎症作用を示すことが複数の論文において報告されている6,7).ところが,一部の研究者はこれまで一定の条件下ではアディポネクチンが炎症を惹起しうることを報告しており8,9),これら矛盾する結果をどのように説明すればよいかは謎であった.この研究では未知のアディポネクチン受容体の存在が示唆され,この受容体が炎症性細胞であるマクロファージにおいてNF-κBを介してインターロイキン6の発現誘導にかかわることが明らかになった.もし,この未知の受容体を含めたアディポネクチンの複数の受容体が環境あるいは細胞に依存して異なる発現パターンをとり異なる(あるいは,逆の)作用を示すものと仮定すると,これまでの相反する結果を矛盾なく説明できるであろう.
そして,インターロイキン6の作用についてもまたいくつかの相反する報告がなされてきた.すなわち,肥満あるいはインスリン抵抗性の状態においてはインターロイキン6の血中濃度が慢性的に上昇しており,このことはインスリン抵抗性の原因のひとつとなる10).一方で,一部の研究者によりインターロイキン6の投与がインスリン感受性を亢進させる場合があると報告されていたが11),その機序は不明であり,それ以上に,肥満あるいはインスリン抵抗性の状態においてインターロイキン6の上昇していることと矛盾すると反論されてきた.この研究では,インターロイキン6の一過性の上昇がSTAT3の活性化を介して肝臓におけるIRS2の発現を上昇させ,これがインスリン感受性を亢進させることが示された.筆者らは,インターロイキン6の一過性の上昇がSTAT3を活性化することができるならば,ひき続いて肝臓におけるIRS2の発現上昇とインスリン感受性の亢進作用を惹起しうる一方で,慢性的にインターロイキン6の上昇している環境においては,おそらくはSOCS3などの負のフィードバックがはたらいてSTAT3の活性化は起こらず,むしろ,このSOCS3などがインスリンの作用を阻害しているのではないか,と提唱している.実際に,運動などにより筋肉から一過性にインターロイキン6の分泌されることが知られており12),今回の報告が,インターロイキン6の生理学的な役割についてのより踏み込んだ研究および理解につながる一助となることが期待される.
文 献
- Taniguchi, C. M., Emanuelli, B. & Kahn, C. R.: Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell Biol., 7, 85-96 (2006)[PubMed]
- Hotamisligil, G. S., Peraldi, P., Budavari, A. et al.: IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α- and obesity-induced insulin resistance. Science, 271, 665-670 (1996)[PubMed]
- Shimomura, I., Matsuda, M., Hammer, R. E. et al.: Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol. Cell, 6, 77-86 (2000)[PubMed]
- Kershaw, E. E. & Flier, J. S.: Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab., 89, 2548-2556 (2004)[PubMed]
- Kadowaki, T., Yamauchi, T., Kubota, N. et al.: Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Invest., 116, 1784-1792 (2006)[PubMed]
- Devaraj, S., Torok, N., Dasu, M. R. et al.: Adiponectin decreases C-reactive protein synthesis and secretion from endothelial cells: evidence for an adipose tissue-vascular loop. Arterioscler. Thromb. Vasc. Biol., 28, 1368-1374 (2008)[PubMed]
- Zhang, P., Wang, Y., Fan, Y. et al.: Overexpression of adiponectin receptors potentiates the antiinflammatory action of subeffective dose of globular adiponectin in vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol., 29, 67-74 (2009)[PubMed]
- Haugen, F. & Drevon, C. A.: Activation of nuclear factor-κB by high molecular weight and globular adiponectin. Endocrinology, 148, 5478-5486 (2007)[PubMed]
- Rovin, B. H. & Song, H.: Chemokine induction by the adipocyte-derived cytokine adiponectin. Clin. Immunol., 120, 99-105 (2007)[PubMed]
- Pradhan, A. D., Manson, J. E., Rifai, N. et al.: C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA, 286, 327-334 (2001)[PubMed]
- Pedersen, B. K. & Febbraio, M. A.: Muscle as an endocrine organ: focus on muscle-derived interleukin-6. Physiol. Rev., 88, 1379-1406 (2008)[PubMed]
- Febbraio, M. A., Hiscock, N., Sacchetti, M. et al.: Interleukin-6 is a novel factor mediating glucose homeostasis during skeletal muscle contraction. Diabetes, 53, 1643-1648 (2004)[PubMed]
著者プロフィール
略歴:2007年 東京大学大学院医学系研究科 修了,2008年 国立健康・栄養研究所 特任研究員を経て,2009年より東京大学大学院医学系研究科 助教.
研究テーマ:アディポカインの生理学,膵β細胞における小胞体ストレス応答系.
関心事:ヒトの摂食行動を分子生物学的にどこまで説明できるか.
植木 浩二郎(Kohjiro Ueki)
東京大学大学院医学系研究科 准教授.
門脇 孝(Takashi Kadowaki)
東京大学大学院医学系研究科 教授.
© 2011 粟澤元晴・植木浩二郎・門脇 孝 Licensed under CC 表示 2.1 日本
