SCFFBW7ユビキチンリガーゼ複合体はアポトーシス抑制タンパク質MCL1をプロテアソーム依存的に分解することにより細胞死を制御する
犬塚博之・Wenyi Wei
(米国Harvard大学Medical School,Beth Israel Deaconess Medical Center,Department of Pathology)
email:犬塚博之
DOI: 10.7875/first.author.2011.060
SCFFBW7 regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction.
Hiroyuki Inuzuka, Shavali Shaik, Ichiro Onoyama, Daming Gao, Alan Tseng, Richard S. Maser, Bo Zhai, Lixin Wan, Alejandro Gutierrez, Alan W. Lau, Yonghong Xiao, Amanda L. Christie, Jon Aster, Jeffrey Settleman, Steven P. Gygi, Andrew L. Kung, Thomas Look, Keiichi I. Nakayama, Ronald A. DePinho, Wenyi Wei
Nature, 471, 104-109 (2011)
細胞の増殖,分化,アポトーシスや代謝などさまざまな細胞機能の調節においてユビキチン-プロテアソーム系を介したタンパク質分解制御機構が重要な役割を担っており,それら制御機構の破綻ががんをはじめとするさまざまな疾患の要因ともなっている.SCFFBW7ユビキチンリガーゼ複合体の基質認識サブユニットFBW7は,ある種の細胞増殖促進タンパク質と選択的に結合することによりそれらのプロテアソーム依存的な分解を促進する.多くのがんにおいてFBW7遺伝子の変異あるいは欠失がみつかっており,FBW7遺伝子のがん抑制遺伝子としての重要性が認識されるなか,SCFFBW7ユビキチンリガーゼ複合体の新たな基質の同定は分子標的療法における標的タンパク質の同定という観点からも重要な意味をもつものと考えられる.筆者らは,T細胞性急性リンパ性白血病において機能欠失型FBW7変異と相関してアポトーシス抑制タンパク質MCL1が過剰発現していることを見い出し,さらに,SCFFBW7ユビキチンリガーゼ複合体によるMCL1のユビキチン-プロテアソーム系を介した分解の分子機構を明らかにした.今回の報告では,MCL1の分解を介したアポトーシスの制御というFBW7の新たながん抑制機能のほか,FBW7変異がT細胞性急性リンパ性白血病に対する分子標的療法における薬剤および患者の選択において有効な指標となりうるという知見を提示することができた.
SCFユビキチンリガーゼ複合体はSkp1,Cul1,Rbx1,Fボックスタンパク質から構成され,そのうちFボックスタンパク質は可変型の基質認識サブユニットとして機能している.SCFユビキチンリガーゼ複合体はおよそ70種類のFボックスタンパク質を選択的に使い分けることにより基質認識の多様性や基質選択の特異性を生み出している1,2).なかでも,FBW7をFボックスタンパク質としてもつSCFFBW7ユビキチンリガーゼ複合体は,細胞周期や,染色体の安定性,幹細胞の維持,脂質代謝の調節など,その多彩な機能が報告されてきており,もっとも注目されるユビキチンリガーゼ複合体のひとつとなっている.
SCFFBW7ユビキチンリガーゼ複合体が分解の役割を担うおもな標的タンパク質としてはサイクリンE,c-Myc,c-Jun,Notch,mTORなど細胞増殖や生存に重要とされるものが多く報告されている2,3).さらに,Fbw7の遺伝子改変マウスの解析において,Fbw7ヘテロノックアウトマウスは放射線への感受性によりリンパ腫を誘発するほか2),T細胞および骨髄に特異的なFbw7ノックアウトマウスの表現型として胸腺リンパ腫および白血病の発症が観察されている4,5).以上のことから,FBW7遺伝子は主要ながん抑制遺伝子のひとつとして位置づけられている.
FBW7遺伝子はさまざまながんでその変異あるいは欠失がみつかっており,とくに,T細胞性急性リンパ性白血病(T-cell acute lymphoblastic leukemia,T-ALL)においてその腫瘍サンプルの約30%でFBW7遺伝子の変異が報告されている3,6).興味深いことに,これら変異の約半数はFBW7の基質認識ドメイン(WD40リピート)にある特定のArg残基(Arg465,Arg479,Arg505)の置換をともなったものである.さらに,これら遺伝学的な知見にくわえ,骨髄に特異的なFbw7ノックアウトマウスのほとんどがT細胞性急性リンパ性白血病を発症することからも5),FBW7がT細胞性急性リンパ性白血病の発症機構の解明の鍵をにぎる重要ながん抑制タンパク質のひとつであることが示唆されている.FBW7遺伝子が変異したT細胞性急性リンパ性白血病においては,FBW7の標的タンパク質であるc-Myc,c-Jun,Notch1の分解異常による過剰発現がその発症の一因であるものと考えられている.しかしその一方で,これらがん原遺伝子産物の過剰発現が細胞周期の停止もしくは細胞死誘導シグナルの活性化により細胞をアポトーシスへと導くことも知られている.筆者らは,このアポトーシスシグナルを抑制する強力な生存シグナルがFBW7の機能欠失によって誘起されることで腫瘍を悪性化に導いているのではないかという仮説をたてた.この研究では,FBW7の機能を欠失したT細胞性急性リンパ性白血病においてアポトーシス抑制シグナルの恒常的な活性化をひき起こすSCFFBW7ユビキチンリガーゼ複合体の未同定の基質を明らかにし,FBW7変異が関与するT細胞性急性リンパ性白血病の発症機構の一端を解明することを目的とした.
FBW7が基質と結合するためには,基質となるタンパク質がコンセンサスなFBW7認識配列をもっていること,そして,その認識配列のSer残基およびThr残基がリン酸化されていることが必要である3).SCFFBW7ユビキチンリガーゼ複合体の基質の探索をはじめるにあたり,まず,データベースよりFBW7認識配列およびその類似配列を含むタンパク質を網羅的に抽出し,それら候補タンパク質から細胞の増殖や生存にかかわるタンパク質の選定を行った.つぎに,SCFFBW7ユビキチンリガーゼ複合体の個々の構成タンパク質をノックダウンした細胞において,それら選定されたタンパク質の発現が上昇しているかどうかを指標としてスクリーニングを行った.その結果,SCFFBW7ユビキチンリガーゼ複合体の基質候補タンパク質としてアポトーシス抑制タンパク質MCL1を同定した.
MCL1はさまざまな血液悪性腫瘍において過剰な発現が認められている7).その構造からMCL1はアポトーシスの制御に関与するBCL2ファミリーのうちアポトーシス抑制にはたらくBCL2サブファミリーに属しているが,ほかのメンバーとは異なり,N末端領域に長いタンパク質不安定化領域(PEST配列)をもちタンパク質の半減期がきわめて短いという特徴をもっている.MCL1はその不安定性から細胞がさまざまなストレスに対しすみやかに生存か死かを選択する際のスイッチの役割を担っているものと考えられている7).したがって,MCL1の発現制御機構の解析は腫瘍悪性化の分子機構の解明,および,がん細胞の効果的な細胞死誘導を考えるうえで重要な知見をあたえるものと思われた.
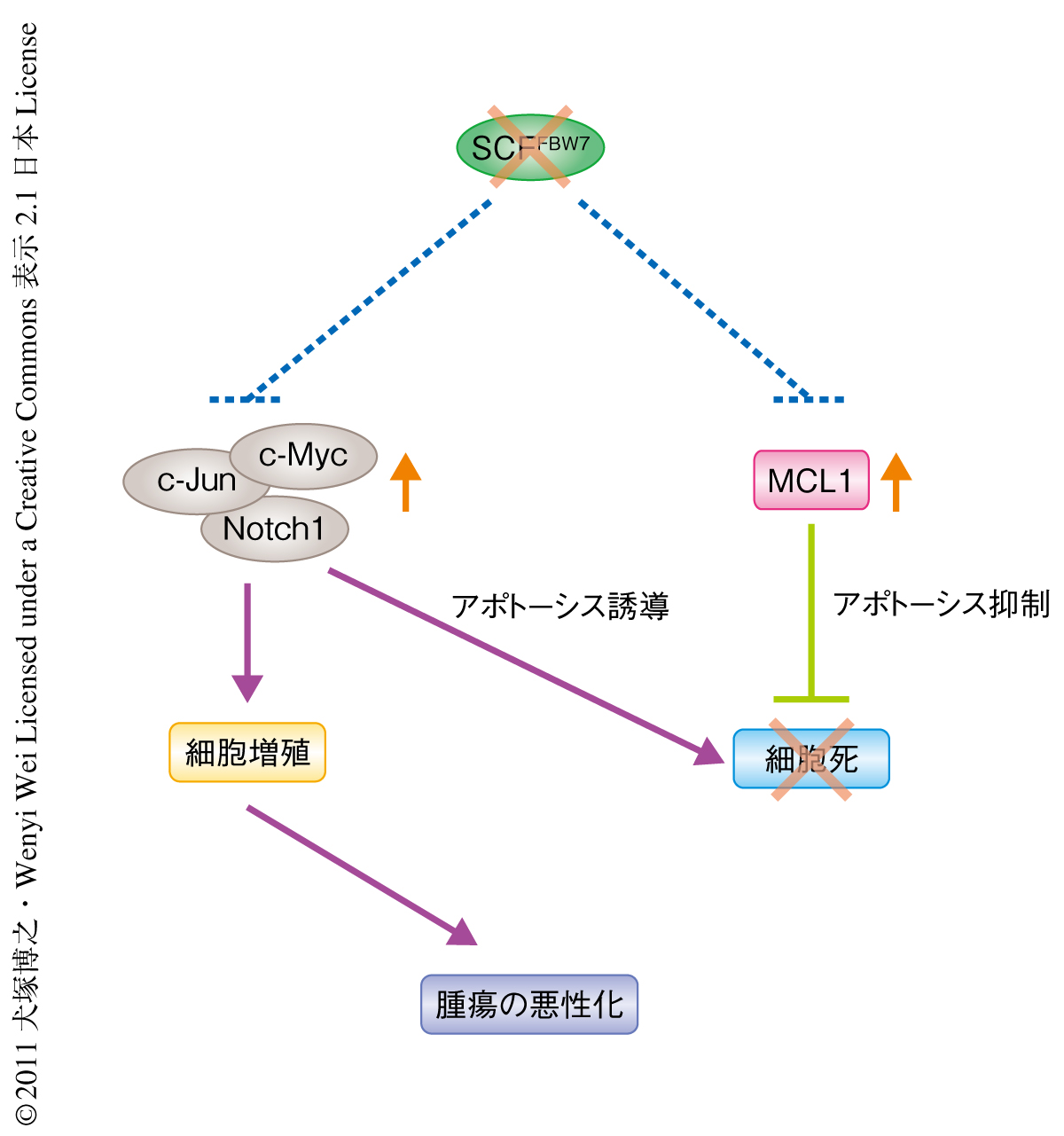
T細胞性急性リンパ性白血病においてMCL1がSCFFBW7ユビキチンリガーゼ複合体の生理的な基質であることを証明するにあたり,はじめに,ヒトT細胞性急性リンパ性白血病細胞株において野生型細胞と機能欠失型FBW7変異細胞とのあいだでMCL1の発現量の比較を行った.その結果,FBW7変異細胞において明らかなMCL1の発現亢進を認めた.野生型細胞においてFBW7をノックダウンするとMCL1の発現が上昇し,逆に,FBW7変異細胞に野生型のFBW7を過剰に発現させるとMCL1の発現量が減少することから,T細胞性急性リンパ性白血病細胞株におけるMCL1の量的な制御はSCFFBW7ユビキチンリガーゼ複合体の活性により直接的に行われていることが示唆された.さらに,培養細胞株だけでなくヒトおよびマウスのT細胞性急性リンパ性白血病の腫瘍サンプルにおいてもFBW7の機能欠失とMCL1の発現量とのあいだに有意な相関を認めた.くわえて,機能欠失型FBW7変異ヒトT細胞性急性リンパ性白血病細胞株をSCIDマウスの尾静脈に注入して作製したヒト化白血病マウスの解析において,過剰に発現するMCL1がマウス生体におけるT細胞性急性リンパ性白血病細胞の増殖を強力に促進することを実験的に証明した.以上の解析結果は,FBW7変異にともなうSCFFBW7ユビキチンリガーゼ複合体の機能欠失がMCL1の細胞における異常な蓄積をひき起こし,T細胞性急性リンパ性白血病の発症およびその腫瘍悪性化に深く関与していることを示唆した(図1).
MCL1がSCFFBW7ユビキチンリガーゼ複合体の基質であることを示すには,FBW7によるMCL1の認識についてその分子機構の詳細な解析を行い,さらに,SCFFBW7ユビキチンリガーゼ複合体によるMCL1へのユビキチン鎖の付加を証明する必要があった.MCL1の安定性はキナーゼGSK3によるリン酸化によって制御されることが報告されていたが8),生理的な条件下でリン酸化型のMCL1を標的とするユビキチンリガーゼはみつかっていなかった.これまでの多くの研究で,FBW7が基質を認識するための基質側の修飾にGSK3の活性が重要であることが報告されていたことから3),SCFFBW7ユビキチンリガーゼ複合体によるMCL1の分解制御を仮定するにあたり,やはり,GSK3がその役割を担いMCL1へのFBW7の結合を誘導しているのではないかと推測した.
これを証明するにあたり,まず,質量分析法およびGSK3のin vitroリン酸化アッセイ法を用いてMCL1のGSK3によるリン酸化部位の同定を行った.そして,どのリン酸化残基がFBW7により認識されるかを調べるため,GSK3によりリン酸化をうけるSer残基およびThr残基をAla残基に置換したMCL1変異体を用いて,FBW7とMCL1との相互作用を試験管内および培養細胞において観察した.その結果,MCL1のN末端側にあるPEST配列のSer159およびThr163がFBW7によるMCL1の結合に必須であることが明らかにされた.さらに,細胞においてGSK3をFBW7とともに強制発現させるとMCL1の半減期が有意に減少するほか,SCFFBW7ユビキチンリガーゼ複合体がGSK3によるMCL1のリン酸化に依存してMCL1をポリユビキチン化することを示すことができた.以上の解析結果から,GSK3によるMCL1のリン酸化がFBW7のMCL1不安定化領域への結合を誘導し,SCFFBW7ユビキチンリガーゼ複合体によるMCL1のユビキチン化とそれにつづくプロテアソーム依存的な分解を促進するものと結論づけた.
T細胞性急性リンパ性白血病の腫瘍サンプルにおいて高頻度にFBW7変異が認められることから,SCFFBW7ユビキチンリガーゼ複合体によるMCL1の分解制御の役割を臨床学的に考察しそれらを薬理学的なアプローチにつなげることがT細胞性急性リンパ性白血病に対する効果的な化学療法の確立という点でも重要であると考えた.この解析では,MCL1の活性を指標とした細胞死の検出を目的とし,アポトーシス抑制タンパク質BCL2サブファミリーの阻害剤である低分子化合物ABT-737に着目し実験に用いた.ABT-737はBCL2ファミリーのうちアポトーシス促進タンパク質であるBH3-onlyサブファミリーの活性を模倣したBH3模倣化合物で,アポトーシス研究分野における最初の分子標的医薬である9).BCL2サブファミリーのうちBCL2,BCLxL,BCLwのBH3ドメイン結合ポケットへの高親和性結合能をもち(阻害定数は1 nM以下),これらの抗アポトーシス機能を強力に抑制することにより,BCL2ファミリーのなかでアポトーシスを促進するタイプのサブファミリーのメンバーの機能を亢進させ細胞死を誘導する.このような作用機序によりABT-737は血液悪性腫瘍や小細胞肺がんに対し単独の投与で高い抗腫瘍効果を示すことが知られている9).その一方で,同じBCL2サブファミリーのメンバーであるMCL1に対する親和性はきわめて低く(阻害定数は1μM以上),MCL1を過剰発現する多くの腫瘍においてABT-737に対する耐性が報告されている9).
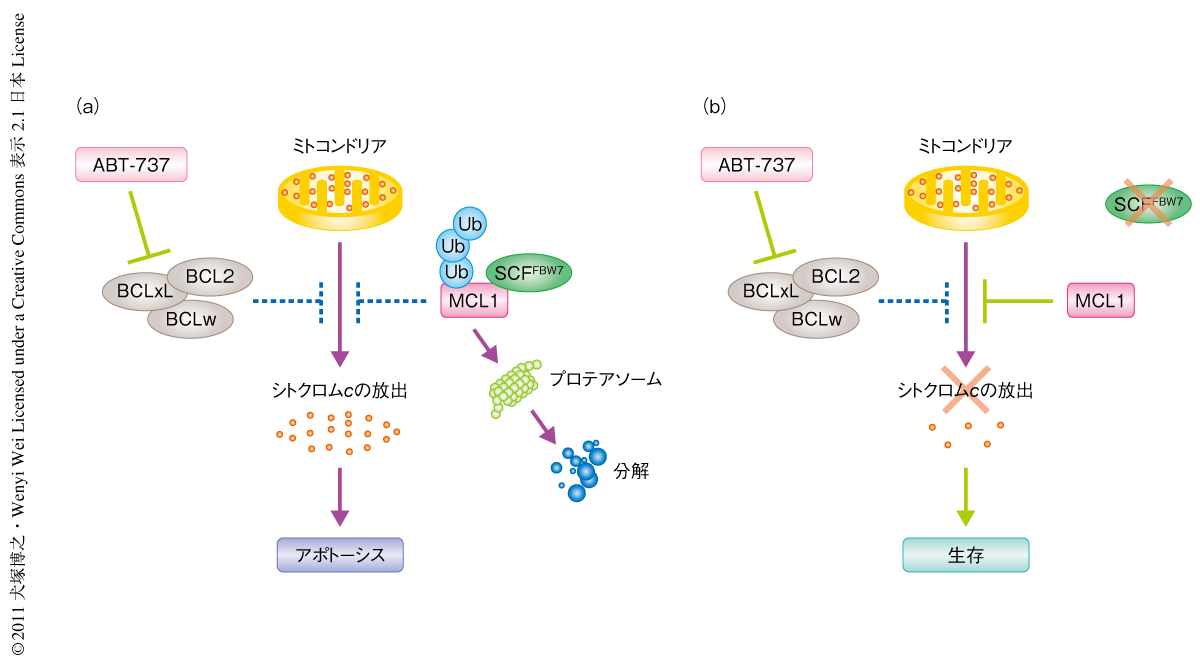
これらの知見をふまえ,T細胞性急性リンパ性白血病細胞株において機能欠失型FBW7変異がABT-737感受性にどのような影響をあたえるかを検討した.野生型およびFBW7変異型のT細胞性急性リンパ性白血病細胞をABT-737で処理し細胞生存率およびアポトーシス誘導率を測定したところ,野生型細胞で顕著なアポトーシス誘導が検出された一方,MCL1を過剰発現する機能欠失型FBW7変異細胞では明らかなABT-737耐性が観察された(図2).機能欠失型FBW7変異細胞において過剰発現するMCL1をノックダウンすることによりABT-737感受性が明らかに回復したことから,野生型細胞とFBW7変異細胞とのあいだでのABT-737感受性の差異はMCL1の発現量により明確に規定されていることになった.したがって,細胞に異常蓄積したMCL1を標的とし選択性をもつ薬理学的な阻害により細胞に効果的にアポトーシスを誘導するという分子標的の考えから,実際にMCL1の発現抑制効果をもつ抗腫瘍薬Sorafenib 7) をABT-737と併用して細胞に処理したところ,機能欠失型FBW7変異T細胞性急性リンパ性白血病細胞においてこれら薬剤による相乗的なアポトーシスを誘導することができた.以上の解析により示されたデータは,FBW7変異を指標としたT細胞性急性リンパ性白血病に対する併用療法の可能性を考えるうえで興味深い結果であると思われた.
今回の解析により,がん抑制タンパク質FBW7を基質認識サブユニットとするSCFFBW7ユビキチンリガーゼ複合体の新たな基質としてアポトーシス抑制タンパク質MCL1が同定された.ヒトT細胞性急性リンパ性白血病細胞株において機能欠失型FBW7変異とMCL1の過剰発現とのあいだに明らかな相関を見い出し,さらに,ヒト化白血病モデルマウスを用いた解析から細胞に異常に蓄積したMCL1が生体におけるT細胞性急性リンパ性白血病細胞の増殖の強力な促進タンパク質となっていることが証明された.
さまざまな血液悪性腫瘍や固形腫瘍においてMCL1の過剰な発現が報告されておりMCL1阻害剤の開発が進められている7).今回の解析において,FBW7遺伝子の変異あるいは欠失によるMCL1の過剰な発現がヒトT細胞性急性リンパ性白血病細胞においてBCL2の阻害剤ABT-737に対し耐性を付与すること,そして,FBW7の機能を欠失したT細胞性急性リンパ性白血病細胞に対するMCL1阻害剤SorafenibとABT-737の併用処理が高い細胞死の誘導効果を示すことがわかった.Nature誌においてこの論文と同じ号に掲載された別のグループの報告からも,卵巣がんをはじめとするさまざまな固形腫瘍細胞株においてFBW7変異によるMCL1の分解異常ががん治療薬である抗チューブリン薬に対し耐性をもたらすことが明らかにされている10).
分子標的療法の治療精度の向上は,高い投薬効果を期待できる薬剤または患者を的確に同定することを可能にする診断法の確立に大きく依存している.T細胞性急性リンパ性白血病をはじめさまざまな悪性腫瘍に対する今後の個別治療の確立にあたり,FBW7変異が分子標的療法における有効な指標となる可能性を示唆できたことは意義深いことである.
略歴:2000年 東京大学大学院農学生命科学研究科博士課程 修了,同年 千葉県がんセンター 研修生,2001年 香川医科大学医学部 教務職員,2004年 米国Emory大学School of Medicine 研究員,2006年 米国Harvard大学Medical School 研究員を経て,2010年より同 インストラクター.
研究テーマ:SCFユビキチンリガーゼ複合体の生理機能と発がん.
抱負:ユビキチン-プロテアソーム系による多彩な生命現象を明らかにしていくことにより,タンパク質分解経路の異常によって発症する疾患の分子機序の解明,および,その治療法の開発にたずさわっていきたい.
Wenyi Wei
米国Harvard大学Medical SchoolにてAssistant Professor.
© 2011 犬塚博之・Wenyi Wei Licensed under CC 表示 2.1 日本
(米国Harvard大学Medical School,Beth Israel Deaconess Medical Center,Department of Pathology)
email:犬塚博之
DOI: 10.7875/first.author.2011.060
SCFFBW7 regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction.
Hiroyuki Inuzuka, Shavali Shaik, Ichiro Onoyama, Daming Gao, Alan Tseng, Richard S. Maser, Bo Zhai, Lixin Wan, Alejandro Gutierrez, Alan W. Lau, Yonghong Xiao, Amanda L. Christie, Jon Aster, Jeffrey Settleman, Steven P. Gygi, Andrew L. Kung, Thomas Look, Keiichi I. Nakayama, Ronald A. DePinho, Wenyi Wei
Nature, 471, 104-109 (2011)
要 約
細胞の増殖,分化,アポトーシスや代謝などさまざまな細胞機能の調節においてユビキチン-プロテアソーム系を介したタンパク質分解制御機構が重要な役割を担っており,それら制御機構の破綻ががんをはじめとするさまざまな疾患の要因ともなっている.SCFFBW7ユビキチンリガーゼ複合体の基質認識サブユニットFBW7は,ある種の細胞増殖促進タンパク質と選択的に結合することによりそれらのプロテアソーム依存的な分解を促進する.多くのがんにおいてFBW7遺伝子の変異あるいは欠失がみつかっており,FBW7遺伝子のがん抑制遺伝子としての重要性が認識されるなか,SCFFBW7ユビキチンリガーゼ複合体の新たな基質の同定は分子標的療法における標的タンパク質の同定という観点からも重要な意味をもつものと考えられる.筆者らは,T細胞性急性リンパ性白血病において機能欠失型FBW7変異と相関してアポトーシス抑制タンパク質MCL1が過剰発現していることを見い出し,さらに,SCFFBW7ユビキチンリガーゼ複合体によるMCL1のユビキチン-プロテアソーム系を介した分解の分子機構を明らかにした.今回の報告では,MCL1の分解を介したアポトーシスの制御というFBW7の新たながん抑制機能のほか,FBW7変異がT細胞性急性リンパ性白血病に対する分子標的療法における薬剤および患者の選択において有効な指標となりうるという知見を提示することができた.
はじめに
SCFユビキチンリガーゼ複合体はSkp1,Cul1,Rbx1,Fボックスタンパク質から構成され,そのうちFボックスタンパク質は可変型の基質認識サブユニットとして機能している.SCFユビキチンリガーゼ複合体はおよそ70種類のFボックスタンパク質を選択的に使い分けることにより基質認識の多様性や基質選択の特異性を生み出している1,2).なかでも,FBW7をFボックスタンパク質としてもつSCFFBW7ユビキチンリガーゼ複合体は,細胞周期や,染色体の安定性,幹細胞の維持,脂質代謝の調節など,その多彩な機能が報告されてきており,もっとも注目されるユビキチンリガーゼ複合体のひとつとなっている.
SCFFBW7ユビキチンリガーゼ複合体が分解の役割を担うおもな標的タンパク質としてはサイクリンE,c-Myc,c-Jun,Notch,mTORなど細胞増殖や生存に重要とされるものが多く報告されている2,3).さらに,Fbw7の遺伝子改変マウスの解析において,Fbw7ヘテロノックアウトマウスは放射線への感受性によりリンパ腫を誘発するほか2),T細胞および骨髄に特異的なFbw7ノックアウトマウスの表現型として胸腺リンパ腫および白血病の発症が観察されている4,5).以上のことから,FBW7遺伝子は主要ながん抑制遺伝子のひとつとして位置づけられている.
FBW7遺伝子はさまざまながんでその変異あるいは欠失がみつかっており,とくに,T細胞性急性リンパ性白血病(T-cell acute lymphoblastic leukemia,T-ALL)においてその腫瘍サンプルの約30%でFBW7遺伝子の変異が報告されている3,6).興味深いことに,これら変異の約半数はFBW7の基質認識ドメイン(WD40リピート)にある特定のArg残基(Arg465,Arg479,Arg505)の置換をともなったものである.さらに,これら遺伝学的な知見にくわえ,骨髄に特異的なFbw7ノックアウトマウスのほとんどがT細胞性急性リンパ性白血病を発症することからも5),FBW7がT細胞性急性リンパ性白血病の発症機構の解明の鍵をにぎる重要ながん抑制タンパク質のひとつであることが示唆されている.FBW7遺伝子が変異したT細胞性急性リンパ性白血病においては,FBW7の標的タンパク質であるc-Myc,c-Jun,Notch1の分解異常による過剰発現がその発症の一因であるものと考えられている.しかしその一方で,これらがん原遺伝子産物の過剰発現が細胞周期の停止もしくは細胞死誘導シグナルの活性化により細胞をアポトーシスへと導くことも知られている.筆者らは,このアポトーシスシグナルを抑制する強力な生存シグナルがFBW7の機能欠失によって誘起されることで腫瘍を悪性化に導いているのではないかという仮説をたてた.この研究では,FBW7の機能を欠失したT細胞性急性リンパ性白血病においてアポトーシス抑制シグナルの恒常的な活性化をひき起こすSCFFBW7ユビキチンリガーゼ複合体の未同定の基質を明らかにし,FBW7変異が関与するT細胞性急性リンパ性白血病の発症機構の一端を解明することを目的とした.
1.T細胞性急性リンパ性白血病細胞株において機能欠失型FBW7変異によりMCL1が細胞に蓄積する
FBW7が基質と結合するためには,基質となるタンパク質がコンセンサスなFBW7認識配列をもっていること,そして,その認識配列のSer残基およびThr残基がリン酸化されていることが必要である3).SCFFBW7ユビキチンリガーゼ複合体の基質の探索をはじめるにあたり,まず,データベースよりFBW7認識配列およびその類似配列を含むタンパク質を網羅的に抽出し,それら候補タンパク質から細胞の増殖や生存にかかわるタンパク質の選定を行った.つぎに,SCFFBW7ユビキチンリガーゼ複合体の個々の構成タンパク質をノックダウンした細胞において,それら選定されたタンパク質の発現が上昇しているかどうかを指標としてスクリーニングを行った.その結果,SCFFBW7ユビキチンリガーゼ複合体の基質候補タンパク質としてアポトーシス抑制タンパク質MCL1を同定した.
MCL1はさまざまな血液悪性腫瘍において過剰な発現が認められている7).その構造からMCL1はアポトーシスの制御に関与するBCL2ファミリーのうちアポトーシス抑制にはたらくBCL2サブファミリーに属しているが,ほかのメンバーとは異なり,N末端領域に長いタンパク質不安定化領域(PEST配列)をもちタンパク質の半減期がきわめて短いという特徴をもっている.MCL1はその不安定性から細胞がさまざまなストレスに対しすみやかに生存か死かを選択する際のスイッチの役割を担っているものと考えられている7).したがって,MCL1の発現制御機構の解析は腫瘍悪性化の分子機構の解明,および,がん細胞の効果的な細胞死誘導を考えるうえで重要な知見をあたえるものと思われた.
T細胞性急性リンパ性白血病においてMCL1がSCFFBW7ユビキチンリガーゼ複合体の生理的な基質であることを証明するにあたり,はじめに,ヒトT細胞性急性リンパ性白血病細胞株において野生型細胞と機能欠失型FBW7変異細胞とのあいだでMCL1の発現量の比較を行った.その結果,FBW7変異細胞において明らかなMCL1の発現亢進を認めた.野生型細胞においてFBW7をノックダウンするとMCL1の発現が上昇し,逆に,FBW7変異細胞に野生型のFBW7を過剰に発現させるとMCL1の発現量が減少することから,T細胞性急性リンパ性白血病細胞株におけるMCL1の量的な制御はSCFFBW7ユビキチンリガーゼ複合体の活性により直接的に行われていることが示唆された.さらに,培養細胞株だけでなくヒトおよびマウスのT細胞性急性リンパ性白血病の腫瘍サンプルにおいてもFBW7の機能欠失とMCL1の発現量とのあいだに有意な相関を認めた.くわえて,機能欠失型FBW7変異ヒトT細胞性急性リンパ性白血病細胞株をSCIDマウスの尾静脈に注入して作製したヒト化白血病マウスの解析において,過剰に発現するMCL1がマウス生体におけるT細胞性急性リンパ性白血病細胞の増殖を強力に促進することを実験的に証明した.以上の解析結果は,FBW7変異にともなうSCFFBW7ユビキチンリガーゼ複合体の機能欠失がMCL1の細胞における異常な蓄積をひき起こし,T細胞性急性リンパ性白血病の発症およびその腫瘍悪性化に深く関与していることを示唆した(図1).
2.SCFFBW7ユビキチンリガーゼ複合体はリン酸化依存的にMCL1をユビキチン化する
MCL1がSCFFBW7ユビキチンリガーゼ複合体の基質であることを示すには,FBW7によるMCL1の認識についてその分子機構の詳細な解析を行い,さらに,SCFFBW7ユビキチンリガーゼ複合体によるMCL1へのユビキチン鎖の付加を証明する必要があった.MCL1の安定性はキナーゼGSK3によるリン酸化によって制御されることが報告されていたが8),生理的な条件下でリン酸化型のMCL1を標的とするユビキチンリガーゼはみつかっていなかった.これまでの多くの研究で,FBW7が基質を認識するための基質側の修飾にGSK3の活性が重要であることが報告されていたことから3),SCFFBW7ユビキチンリガーゼ複合体によるMCL1の分解制御を仮定するにあたり,やはり,GSK3がその役割を担いMCL1へのFBW7の結合を誘導しているのではないかと推測した.
これを証明するにあたり,まず,質量分析法およびGSK3のin vitroリン酸化アッセイ法を用いてMCL1のGSK3によるリン酸化部位の同定を行った.そして,どのリン酸化残基がFBW7により認識されるかを調べるため,GSK3によりリン酸化をうけるSer残基およびThr残基をAla残基に置換したMCL1変異体を用いて,FBW7とMCL1との相互作用を試験管内および培養細胞において観察した.その結果,MCL1のN末端側にあるPEST配列のSer159およびThr163がFBW7によるMCL1の結合に必須であることが明らかにされた.さらに,細胞においてGSK3をFBW7とともに強制発現させるとMCL1の半減期が有意に減少するほか,SCFFBW7ユビキチンリガーゼ複合体がGSK3によるMCL1のリン酸化に依存してMCL1をポリユビキチン化することを示すことができた.以上の解析結果から,GSK3によるMCL1のリン酸化がFBW7のMCL1不安定化領域への結合を誘導し,SCFFBW7ユビキチンリガーゼ複合体によるMCL1のユビキチン化とそれにつづくプロテアソーム依存的な分解を促進するものと結論づけた.
3.SCFFBW7ユビキチンリガーゼ複合体はMCL1の分解を介して細胞死を制御する
T細胞性急性リンパ性白血病の腫瘍サンプルにおいて高頻度にFBW7変異が認められることから,SCFFBW7ユビキチンリガーゼ複合体によるMCL1の分解制御の役割を臨床学的に考察しそれらを薬理学的なアプローチにつなげることがT細胞性急性リンパ性白血病に対する効果的な化学療法の確立という点でも重要であると考えた.この解析では,MCL1の活性を指標とした細胞死の検出を目的とし,アポトーシス抑制タンパク質BCL2サブファミリーの阻害剤である低分子化合物ABT-737に着目し実験に用いた.ABT-737はBCL2ファミリーのうちアポトーシス促進タンパク質であるBH3-onlyサブファミリーの活性を模倣したBH3模倣化合物で,アポトーシス研究分野における最初の分子標的医薬である9).BCL2サブファミリーのうちBCL2,BCLxL,BCLwのBH3ドメイン結合ポケットへの高親和性結合能をもち(阻害定数は1 nM以下),これらの抗アポトーシス機能を強力に抑制することにより,BCL2ファミリーのなかでアポトーシスを促進するタイプのサブファミリーのメンバーの機能を亢進させ細胞死を誘導する.このような作用機序によりABT-737は血液悪性腫瘍や小細胞肺がんに対し単独の投与で高い抗腫瘍効果を示すことが知られている9).その一方で,同じBCL2サブファミリーのメンバーであるMCL1に対する親和性はきわめて低く(阻害定数は1μM以上),MCL1を過剰発現する多くの腫瘍においてABT-737に対する耐性が報告されている9).
これらの知見をふまえ,T細胞性急性リンパ性白血病細胞株において機能欠失型FBW7変異がABT-737感受性にどのような影響をあたえるかを検討した.野生型およびFBW7変異型のT細胞性急性リンパ性白血病細胞をABT-737で処理し細胞生存率およびアポトーシス誘導率を測定したところ,野生型細胞で顕著なアポトーシス誘導が検出された一方,MCL1を過剰発現する機能欠失型FBW7変異細胞では明らかなABT-737耐性が観察された(図2).機能欠失型FBW7変異細胞において過剰発現するMCL1をノックダウンすることによりABT-737感受性が明らかに回復したことから,野生型細胞とFBW7変異細胞とのあいだでのABT-737感受性の差異はMCL1の発現量により明確に規定されていることになった.したがって,細胞に異常蓄積したMCL1を標的とし選択性をもつ薬理学的な阻害により細胞に効果的にアポトーシスを誘導するという分子標的の考えから,実際にMCL1の発現抑制効果をもつ抗腫瘍薬Sorafenib 7) をABT-737と併用して細胞に処理したところ,機能欠失型FBW7変異T細胞性急性リンパ性白血病細胞においてこれら薬剤による相乗的なアポトーシスを誘導することができた.以上の解析により示されたデータは,FBW7変異を指標としたT細胞性急性リンパ性白血病に対する併用療法の可能性を考えるうえで興味深い結果であると思われた.
おわりに
今回の解析により,がん抑制タンパク質FBW7を基質認識サブユニットとするSCFFBW7ユビキチンリガーゼ複合体の新たな基質としてアポトーシス抑制タンパク質MCL1が同定された.ヒトT細胞性急性リンパ性白血病細胞株において機能欠失型FBW7変異とMCL1の過剰発現とのあいだに明らかな相関を見い出し,さらに,ヒト化白血病モデルマウスを用いた解析から細胞に異常に蓄積したMCL1が生体におけるT細胞性急性リンパ性白血病細胞の増殖の強力な促進タンパク質となっていることが証明された.
さまざまな血液悪性腫瘍や固形腫瘍においてMCL1の過剰な発現が報告されておりMCL1阻害剤の開発が進められている7).今回の解析において,FBW7遺伝子の変異あるいは欠失によるMCL1の過剰な発現がヒトT細胞性急性リンパ性白血病細胞においてBCL2の阻害剤ABT-737に対し耐性を付与すること,そして,FBW7の機能を欠失したT細胞性急性リンパ性白血病細胞に対するMCL1阻害剤SorafenibとABT-737の併用処理が高い細胞死の誘導効果を示すことがわかった.Nature誌においてこの論文と同じ号に掲載された別のグループの報告からも,卵巣がんをはじめとするさまざまな固形腫瘍細胞株においてFBW7変異によるMCL1の分解異常ががん治療薬である抗チューブリン薬に対し耐性をもたらすことが明らかにされている10).
分子標的療法の治療精度の向上は,高い投薬効果を期待できる薬剤または患者を的確に同定することを可能にする診断法の確立に大きく依存している.T細胞性急性リンパ性白血病をはじめさまざまな悪性腫瘍に対する今後の個別治療の確立にあたり,FBW7変異が分子標的療法における有効な指標となる可能性を示唆できたことは意義深いことである.
文 献
- Cardozo, T. & Pagano, M.: The SCF ubiquitin ligase: insights into a molecular machine. Nat. Rev. Mol. Cell Biol., 5, 739-751 (2004)[PubMed]
- Nakayama, K. I. & Nakayama, K.: Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer, 6, 369-381 (2006)[PubMed]
- Welcker, M. & Clurman, B. E.: FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev, Cancer, 2, 83-93 (2008)[PubMed]
- Onoyama, I., Tsunematsu, R., Matsumoto, A. et al.: Conditional inactivation of Fbxw7 impairs cell-cycle exit during T cell differentiation and results in lymphomatogenesis. J. Exp. Med., 204, 2875-2888 (2007)[PubMed]
- Matsuoka, S., Oike, Y., Onoyama, I. et al.: Fbxw7 acts as a critical fail-safe against premature loss of hematopoietic stem cells and development of T-ALL. Genes Dev., 986-991 (2008)[PubMed]
- Maser, R. S., Choudhury, B., Campbell, P. J. et al.: Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature, 447, 966-971 (2007)[PubMed]
- Akgul, C.: Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell. Mol. Life Sci., 66, 1326-1336 (2009)[PubMed]
- Maurer, U., Charvet, C., Wagman, A. S. et al.: Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell, 21, 749-760 (2006)[PubMed]
- Lessene, G., Czabotar, P. E. & Colman, P. M.: BCL-2 family antagonists for cancer therapy. Nat. Rev. Drug Discov., 7, 989-1000 (2008)[PubMed]
- Wertz, I. E., Kusam, S., Lam, C. et al.: Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature, 471, 110-104 (2011)[PubMed]
著者プロフィール
略歴:2000年 東京大学大学院農学生命科学研究科博士課程 修了,同年 千葉県がんセンター 研修生,2001年 香川医科大学医学部 教務職員,2004年 米国Emory大学School of Medicine 研究員,2006年 米国Harvard大学Medical School 研究員を経て,2010年より同 インストラクター.
研究テーマ:SCFユビキチンリガーゼ複合体の生理機能と発がん.
抱負:ユビキチン-プロテアソーム系による多彩な生命現象を明らかにしていくことにより,タンパク質分解経路の異常によって発症する疾患の分子機序の解明,および,その治療法の開発にたずさわっていきたい.
Wenyi Wei
米国Harvard大学Medical SchoolにてAssistant Professor.
© 2011 犬塚博之・Wenyi Wei Licensed under CC 表示 2.1 日本
