血管内皮細胞におけるインスリンシグナルの障害は骨格筋へのインスリンの移行を低下させ骨格筋での糖の取り込みを減少させる
窪田直人1・窪田哲也2・門脇 孝3
(1東京大学システム疾患生命科学による先端医療技術開発拠点,2国立健康・栄養研究所栄養療法プロジェクト,3東京大学大学院医学系研究科 代謝・栄養病態学)
email:窪田直人
DOI: 10.7875/first.author.2011.048
Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle.
Tetsuya Kubota, Naoto Kubota, Hiroki Kumagai, Shinichi Yamaguchi, Hideki Kozono, Takehiro Takahashi, Mariko Inoue, Shinsuke Itoh, Iseki Takamoto, Takayoshi Sasako, Katsuyoshi Kumagai, Tomoko Kawai, Shinji Hashimoto, Tsuneo Kobayashi, Maki Sato, Kumpei Tokuyama, Satoshi Nishimura, Masaki Tsunoda, Tomohiro Ide, Koji Murakami, Tomomi Yamazaki, Osamu Ezaki, Koichi Kawamura, Hirotake Masuda, Masao Moroi, Kaoru Sugi, Yuichi Oike, Hiroaki Shimokawa, Nobuyuki Yanagihara, Masato Tsutsui, Yasuo Terauchi, Kazuyuki Tobe, Ryozo Nagai, Katsuo Kamata, Kenji Inoue, Tatsuhiko Kodama, Kohjiro Ueki, Takashi Kadowaki
Cell Metabolism, 13, 294-307 (2011)
肥満や2型糖尿病において認められる骨格筋インスリン抵抗性の分子機構のひとつとして以前より血中から骨格筋間質へのインスリン移行の低下が指摘されているが,その詳細な分子機構については十分には解明されていなかった.筆者らは,この分子機構に血管内皮細胞におけるインスリンシグナルが重要なはたらきをしているものと考え,血管内皮細胞でのインスリン受容体の主要な基質であるIRS2に着目して解析を行った.血管内皮細胞に特異的なIRS2ノックアウトマウスの血管内皮細胞ではインスリン刺激による内皮型NO合成酵素の活性化が低下し,骨格筋における毛細血管の拡張能の障害および骨格筋間質へのインスリンの移行の低下を認め,骨格筋でのインスリン依存性の糖の取り込みが障害されていた.さらに,プロスタグランジンI2アナログによりインスリン刺激による内皮型NO合成酵素の活性化を回復すると骨格筋における毛細血管の拡張能および骨格筋間質へのインスリンの移行が改善し,骨格筋でのインスリン依存性の糖の取り込みの回復が認められた.また,肥満モデルマウスの血管内皮細胞においてもIRS2の顕著な発現低下がみられ,血管内皮細胞でのインスリン刺激による内皮型NO合成酵素の活性化,毛細血管の拡張能,骨格筋間質へのインスリンの移行の低下が認められ,骨格筋でのインスリン依存性の糖の取り込みが障害されていた.プロスタグランジンI2アナログを用いて血管内皮細胞でのインスリン刺激による内皮型NO合成酵素の活性化を回復すると,毛細血管の拡張能および骨格筋間質へのインスリンの移行の回復にともない,この障害も改善することが明らかになった.以上の結果より,血管内皮細胞におけるインスリンシグナルが骨格筋でのインスリン感受性の調節に重要な役割をはたしており,このインスリンシグナルがインスリン抵抗性改善薬の新規のターゲットとなりうることが明らかになった.
現在,わが国では2型糖尿病が増加の一途をたどっており,最新の国民健康・栄養調査の発表によると糖尿病の強く疑われる人が約890万人,糖尿病の可能性を否定できない人をくわえると約2210万人にものぼるといわれている.日本人は遺伝因子として欧米人に比べてインスリンの分泌能が低いうえ(約1/2ともいわれる),環境因子として急速に進んだ食生活の欧米化や運動不足により肥満や内臓脂肪の蓄積にともなうインスリン抵抗性(インスリンが効きにくい状態)が進んだ結果,相対的なインスリンの作用不足におちいり2型糖尿病の急増にいたっていると考えられている.インスリンの主要な作用臓器として肝臓と骨格筋があげられるが,とくに骨格筋はヒトにおいて最大のグルコースの消費臓器であり,骨格筋におけるインスリン抵抗性は2型糖尿病やメタボリックシンドロームをひき起こし心筋梗塞や脳卒中といった大血管合併症や腎症や網膜症といった細小血管合併症の発症要因となっている1,2).したがって,骨格筋におけるインスリンによる糖の取り込み機構の解明や肥満によるインスリン抵抗性の発症の分子機構を明らかにすることは,糖尿病およびメタボリックシンドロームやその合併症の予防や治療において最大のテーマのひとつになっている.
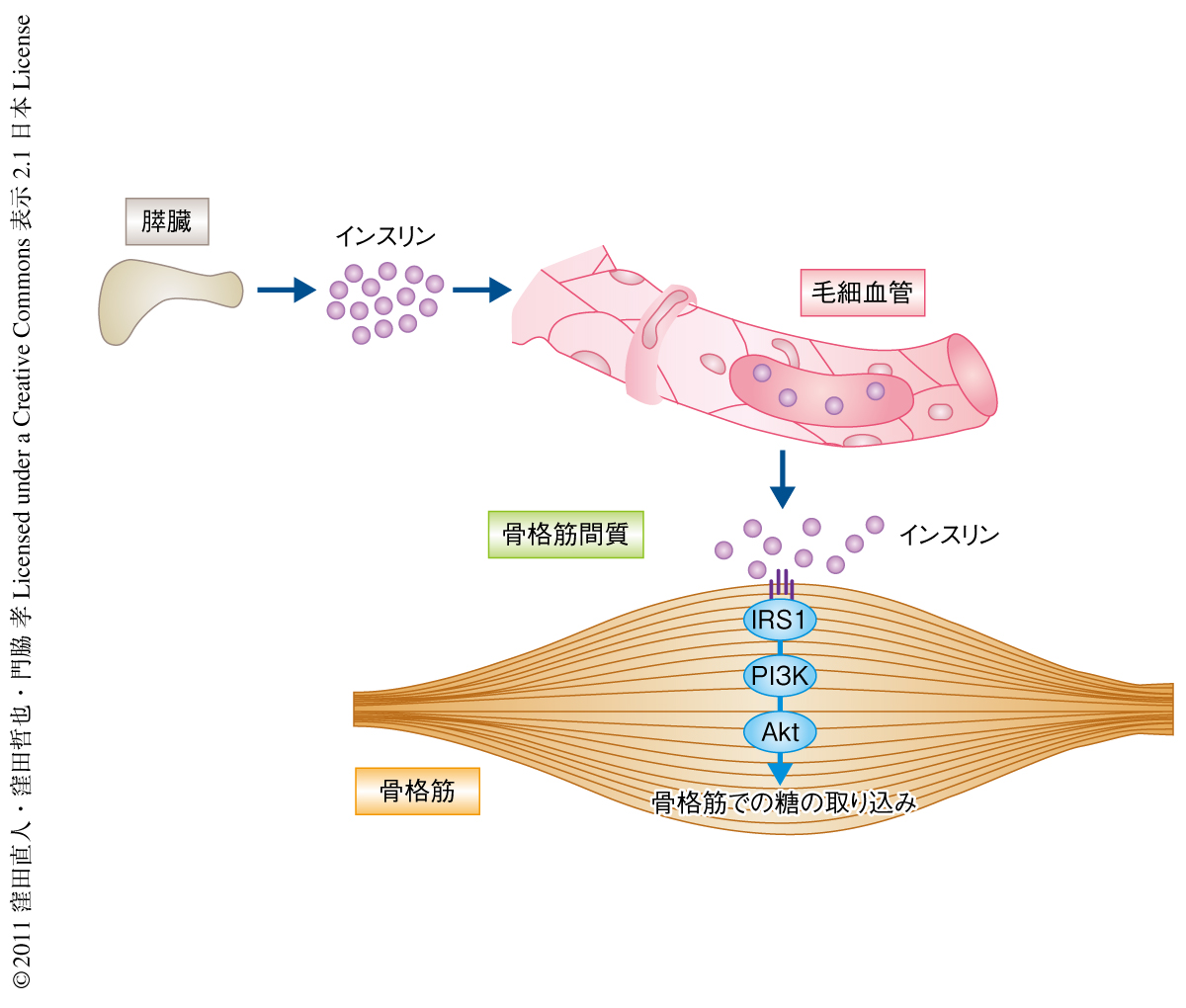
骨格筋においてインスリンが正常に作用するためには,食後に膵臓のランゲルハンス島β細胞から分泌されたインスリンが骨格筋に分布する毛細血管に到達し,毛細血管が拡張して骨格筋間質に移行したインスリンが骨格筋の細胞の表面に存在するインスリン受容体に結合する必要がある3-6)(図1).以前より,骨格筋ではインスリンの移行が血液中や肝臓に比べゆっくりでありインスリン依存性の糖の取り込みも緩徐に起こることが知られている1,7,8).骨格筋細胞を用いたin vitroの実験や骨格筋間質にインスリンを直接に投与したin vivoの実験ではインスリン依存性の糖の取り込みはすみやかに起こることから,骨格筋においてインスリン依存性の糖の取り込みが緩徐な理由はインスリンの移行がゆっくりであるためと理解されている9).さらに,肥満者では血液や肝臓でのインスリンの分布に健常者と差は認められないものの,骨格筋へのインスリンの移行が低下しており,それにともなってインスリン依存性の糖の取り込みが低下していることが知られている10).実際に骨格筋間質のインスリンの濃度を測定した報告では,肥満者ではインスリンの移行速度や濃度が低下している10).こうした一連の知見は,骨格筋ではインスリンの移行が律速段階となりインスリン作用の出現が緩徐となっていること,さらに,肥満にともなう骨格筋のインスリン抵抗性には骨格筋そのものの糖の取り込みの障害にくわえ,生理的に緩徐なインスリンの移行が肥満ではさらに低下しており骨格筋における正常なインスリン依存性の糖の取り込みの障害の一因となっていることを強く示唆する.しかしこれまで,そもそも骨格筋へのインスリンの移行がどのように制御されているのか,そして,肥満ではなぜこれが障害されるのか,その分子機構は十分にわかっていなかった.今回,筆者らは,血管内皮細胞におけるインスリンシグナルが毛細血管の拡張を制御しこれにより骨格筋間質へのインスリンの移行が調節されていること,そして,肥満者では血管内皮細胞のインスリンシグナルの障害がインスリンの移行の低下の原因となっていることを明らかにした.
血管内皮細胞でのインスリン受容体の主要な基質であるIRS2(insulin receptor substrate 2)を血管内皮細胞に特異的に欠損したマウスでは,血管内皮細胞において血管拡張作用をもつNOを産生する酵素である内皮型NO合成酵素のインスリンによる活性化が約1/3~1/4に低下しており,グルコースクランプ法によりインスリン感受性を測定したところ骨格筋においてインスリンによる糖の取り込みが有意に低下していた.単離した骨格筋においてはインスリン依存性および非依存性の糖の取り込みに異常は認められなかったことから,このノックアウトマウスではインスリンの移行が障害されていることが示唆された.まず,マイクロバブルを用いて毛細血管の拡張能について野生型マウスを用いて検討したところインスリンにより有意な拡張が認められ,この拡張は内皮型NO合成酵素の阻害剤であるL-NAMEの投与により完全に阻害されることから内皮型NO合成酵素に依存性であることが確認された.そして,血管内皮細胞に特異的なIRS2ノックアウトマウスではこのインスリンによる毛細血管の拡張が有意に障害されていた.さらに,微小透析(マイクロダイアリシス)プロープを用いて骨格筋間質におけるインスリンの濃度を測定したところ,このノックアウトマウスではインスリンの移行が有意に低下していることが明らかになった.以上の結果から,血管内皮細胞に特異的なIRS2ノックアウトマウスではインスリンによる内皮型NO合成酵素の活性化が十分に起こらないため毛細血管の拡張や骨格筋間質へのインスリンの移行が障害され,骨格筋におけるインスリン依存性の糖の取り込みが低下していることが明らかになった.
血管内皮細胞に特異的なIRS2ノックアウトマウスでは全身の血管内皮細胞においてIRS2が欠損しているにもかかわらず,なぜ肝臓ではインスリン抵抗性が認められずに骨格筋においてのみインスリン抵抗性が認められたのであろうか.毛細血管はその構造から3つのタイプが存在することが知られている.骨格筋や心筋などに分布する閉鎖結合によって連続的に血管内皮細胞が連結された連続性毛細血管と,腎糸球体や内分泌腺,害分泌腺などに分布する血管内皮細胞の薄膜化した部分に大きさの一様な孔のある有窓性(小孔性)毛細血管,そして,肝臓や脾臓に分布する比較的直径が大きくアルブミンや一部の血球成分の行き来が可能な孔と断裂が認められる洞様毛細血管である.おそらく,肝臓ではインスリンはこの孔や断裂をつうじて非常にすみやかに移行することができるのに対し,連続性毛細血管が分布する骨格筋ではインスリンによる毛細血管の拡張がその移行に重要な役割をはたしているため,血管内皮細胞に特異的なIRS2ノックアウトマウスでは骨格筋においてのみインスリン抵抗性が認められたものと思われた.また,さきに述べた,インスリン移行の速度が肝臓では血液中と同じくらいすみやかであるのに対し骨格筋では緩徐であることも,このそれぞれの臓器に分布する毛細血管の構造の違いに密接に関連するものと思われる.
つぎに,血管内皮細胞に特異的なIRS2ノックアウトマウスで認められた表現型をインスリンによる内皮型NO合成酵素の活性化を回復することによりレスキューすることができるかどうか検討した.プロスタグランジンI2アナログはcAMP-プロテインキナーゼA-転写因子CREBを介して血管内皮細胞において内皮型NO合成酵素の発現量を増加させることが報告されている.そこで,このノックアウトマウスにプロスタグランジンI2アナログのひとつであるベラプロストナトリウムを投与し内皮型NO合成酵素の発現量が約2倍となるようにしたところ,血管内皮細胞におけるインスリンによる内皮型NO合成酵素の活性化は野生型マウスとほぼ同じ程度にまで改善し,毛細血管の拡張能も同様に回復した.このベラプロストナトリウムによる毛細血管の拡張能の回復は内皮型NO合成酵素の阻害薬であるL-NAMEにより完全に阻害されたことから内皮型NO合成酵素を介しているものと考えられた.そして,毛細血管の拡張能の回復にともない骨格筋間質へのインスリンの移行もほぼ完全に回復し,骨格筋におけるインスリン依存性の糖の取り込みもほぼ完全に改善した.なお,ベラプロストナトリウムが骨格筋そのものの糖の取り込みに影響をあたえなかったことについては,ベラプロストナトリウムを投与したマウスの骨格筋を単離してインスリン依存性および非依存性の糖の取り込みを検討し,ベラプロストナトリウム非投与群と差が認められないことで確認した.以上の結果から,骨格筋間質へのインスリンの移行とそれにともなうインスリン依存性の骨格筋での糖の取り込みが血管内皮細胞のインスリンシグナル(インスリンによる内皮型NO合成酵素の活性化)により調節されていることが明らかになった.
つぎに,肥満で認められる骨格筋のインスリン抵抗性に同様な分子機構が存在するかどうかについて検討を行った.肥満モデルマウスとして8週間にわたり高脂肪食を負荷したマウスを使用した.高脂肪食負荷マウスの血管内皮細胞ではIRS1の発現が約1/2に,IRS2の発現が1/4に低下しており,それにともなってインスリンによる内皮型NO合成酵素の活性化が血管内皮細胞に特異的なIRS2ノックアウトマウスと同様に約1/3に低下していた.この高脂肪食負荷マウスの毛細血管の拡張能および骨格筋間質へのインスリンの移行を検討したところ,普通食負荷マウスに比べいずれも有意に低下していた.そして,グルコースクランプ法によりインスリン感受性を測定した結果,血管内皮細胞に特異的なIRS2ノックアウトマウスとは異なり肝臓におけるインスリン抵抗性は認められたが,骨格筋におけるインスリン依存性の糖の取り込みも有意に障害されていた.
もし,血管内皮細胞に特異的なIRS2ノックアウトマウスと同じような分子機構でこの骨格筋におけるインスリン抵抗性が生じているのであれば,同様にプロスタグランジンI2アナログであるベラプロストナトリウムによるレスキューが可能なはずである.そこで,さきほどと同様に内皮型NO合成酵素の発現量が約2倍に増加するようベラプロストナトリウムを投与したところ,インスリンによる内皮型NO合成酵素の活性化および毛細血管の拡張能はほぼ普通食負荷マウスと同じ程度にまで改善した.ベラプロストナトリウムによる毛細血管の拡張能の改善作用は内皮型NO合成酵素阻害薬であるL-NAMEにより完全に阻害されたことから内皮型NO合成酵素を介しているものと考えられた.そして,毛細血管の拡張能の改善にともない骨格筋間質へのインスリンの移行もほぼ普通食負荷マウスと同じ程度にまで回復した.グルコースクランプ法により高脂肪食負荷マウスのインスリン抵抗性を測定したところ,肝臓におけるインスリン抵抗性はまったく改善していなかったが骨格筋における糖の取り込みは有意に改善していた.しかし,その改善は毛細血管の拡張能やインスリンの移行とは異なり2/3程度にとどまっていた.これは,ベラプロストナトリウムによりインスリンの移行は改善したが肥満によってもたらされた骨格筋そのもののインスリン作用は改善しなかったためと考えられた.実際に,単離した骨格筋のインスリン依存性および非依存性の糖の取り込みを検討したところ高脂肪食負荷マウスでは普通食負荷マウスに比べ有意に低下しており,これはベラプロストナトリウムを投与してもまったく改善が認められなかった.
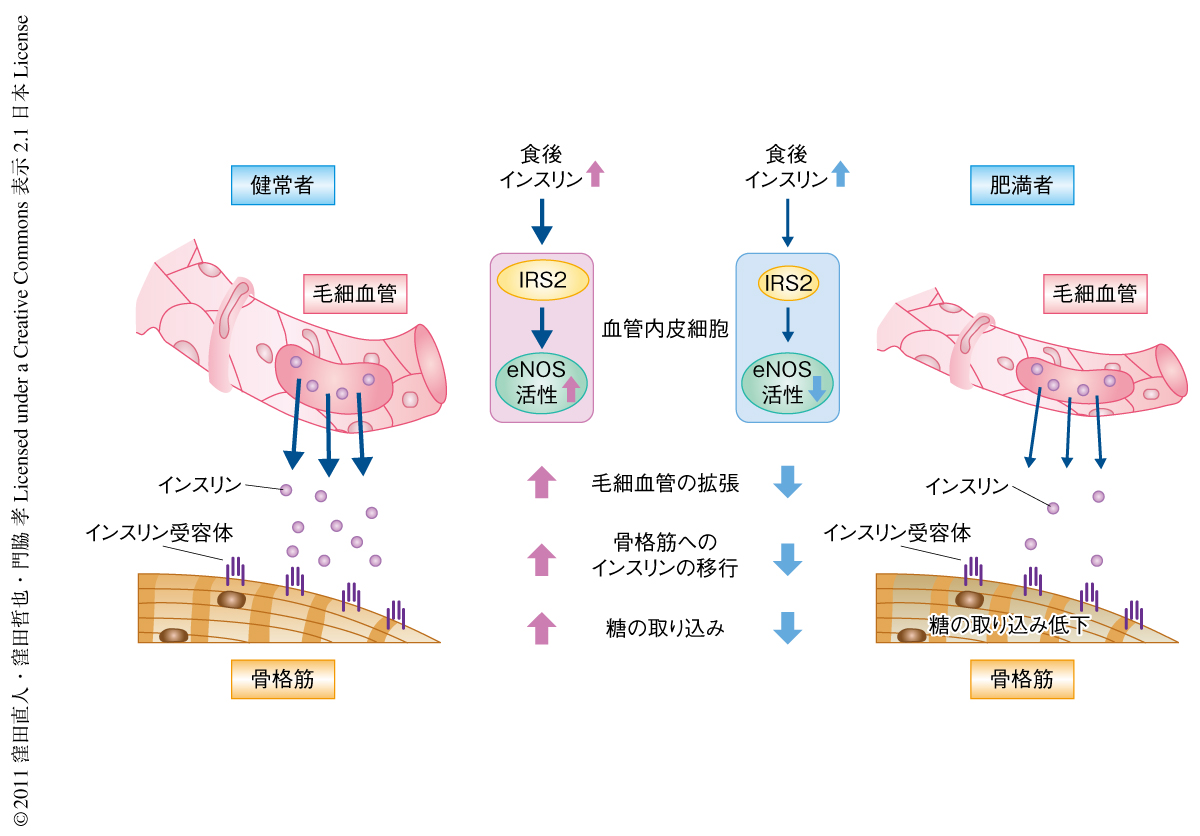
以上の結果より,健常者では食後にインスリンが分泌されると内皮細胞の内皮型NO合成酵素が活性化されて毛細血管が拡張しインスリンが骨格筋間質に移行して正常に糖の取り込みが起こるのに対し,肥満者では血管内皮細胞におけるインスリン受容体基質,とくにIRS2の発現が低下しているため,食後にインスリンが分泌されてもそのシグナルは十分に伝達されずに内皮型NO合成酵素の活性化が低下し,毛細血管の拡張や骨格筋間質へのインスリンの移行が正常に起こらず骨格筋における糖の取り込みが障害されているものと考えられた(図2).そして,プロスタグランジンI2アナログであるベラプロストナトリウムはインスリンによる血管内皮細胞における内皮型NO合成酵素の活性化を改善することで,毛細血管の拡張能および骨格筋間質へのインスリンの移行を改善し骨格筋における糖の取り込みを回復することが明らかになった.これまで糖尿病の治療薬としてさまざまな分子機構をもつ薬剤が臨床で使用されているが,血管内皮細胞をターゲットとした薬剤はいうまでもなく,骨格筋へのインスリンの移行に注目した薬剤もまったく存在しないのが現状である.これまでに登場した糖尿病治療薬ではまだ十分に血糖コントロールのできていない背景の一部に,こうしたまだわれわれがアプローチできていないインスリン抵抗性の分子機構のある可能性は否定できない.今回,使用したベラプロストナトリウムは幸いにも閉塞性動脈硬化症の治療薬としてすでに臨床応用されている.今回の発見が肥満にともなうインスリン抵抗性改善薬の新たな開発につながれば幸いである.
略歴:2001年 東京大学大学院医学系研究科 修了,2004年 東京大学医学部附属病院 客員助教を経て,2007年より東京大学システム疾患生命科学による先端医療技術開発拠点 特任准教授.
研究テーマ:モデル動物を用いた糖尿病・肥満・動脈硬化症の分子機構の解明.
窪田 哲也(Tetsuya Kubota)
国立健康・栄養研究所栄養療法プロジェクト プロジェクトリーダー.
門脇 孝(Takashi Kadowaki)
東京大学大学院医学系研究科 教授.
© 2011 窪田直人・窪田哲也・門脇 孝 Licensed under CC 表示 2.1 日本
(1東京大学システム疾患生命科学による先端医療技術開発拠点,2国立健康・栄養研究所栄養療法プロジェクト,3東京大学大学院医学系研究科 代謝・栄養病態学)
email:窪田直人
DOI: 10.7875/first.author.2011.048
Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle.
Tetsuya Kubota, Naoto Kubota, Hiroki Kumagai, Shinichi Yamaguchi, Hideki Kozono, Takehiro Takahashi, Mariko Inoue, Shinsuke Itoh, Iseki Takamoto, Takayoshi Sasako, Katsuyoshi Kumagai, Tomoko Kawai, Shinji Hashimoto, Tsuneo Kobayashi, Maki Sato, Kumpei Tokuyama, Satoshi Nishimura, Masaki Tsunoda, Tomohiro Ide, Koji Murakami, Tomomi Yamazaki, Osamu Ezaki, Koichi Kawamura, Hirotake Masuda, Masao Moroi, Kaoru Sugi, Yuichi Oike, Hiroaki Shimokawa, Nobuyuki Yanagihara, Masato Tsutsui, Yasuo Terauchi, Kazuyuki Tobe, Ryozo Nagai, Katsuo Kamata, Kenji Inoue, Tatsuhiko Kodama, Kohjiro Ueki, Takashi Kadowaki
Cell Metabolism, 13, 294-307 (2011)
要 約
肥満や2型糖尿病において認められる骨格筋インスリン抵抗性の分子機構のひとつとして以前より血中から骨格筋間質へのインスリン移行の低下が指摘されているが,その詳細な分子機構については十分には解明されていなかった.筆者らは,この分子機構に血管内皮細胞におけるインスリンシグナルが重要なはたらきをしているものと考え,血管内皮細胞でのインスリン受容体の主要な基質であるIRS2に着目して解析を行った.血管内皮細胞に特異的なIRS2ノックアウトマウスの血管内皮細胞ではインスリン刺激による内皮型NO合成酵素の活性化が低下し,骨格筋における毛細血管の拡張能の障害および骨格筋間質へのインスリンの移行の低下を認め,骨格筋でのインスリン依存性の糖の取り込みが障害されていた.さらに,プロスタグランジンI2アナログによりインスリン刺激による内皮型NO合成酵素の活性化を回復すると骨格筋における毛細血管の拡張能および骨格筋間質へのインスリンの移行が改善し,骨格筋でのインスリン依存性の糖の取り込みの回復が認められた.また,肥満モデルマウスの血管内皮細胞においてもIRS2の顕著な発現低下がみられ,血管内皮細胞でのインスリン刺激による内皮型NO合成酵素の活性化,毛細血管の拡張能,骨格筋間質へのインスリンの移行の低下が認められ,骨格筋でのインスリン依存性の糖の取り込みが障害されていた.プロスタグランジンI2アナログを用いて血管内皮細胞でのインスリン刺激による内皮型NO合成酵素の活性化を回復すると,毛細血管の拡張能および骨格筋間質へのインスリンの移行の回復にともない,この障害も改善することが明らかになった.以上の結果より,血管内皮細胞におけるインスリンシグナルが骨格筋でのインスリン感受性の調節に重要な役割をはたしており,このインスリンシグナルがインスリン抵抗性改善薬の新規のターゲットとなりうることが明らかになった.
はじめに
現在,わが国では2型糖尿病が増加の一途をたどっており,最新の国民健康・栄養調査の発表によると糖尿病の強く疑われる人が約890万人,糖尿病の可能性を否定できない人をくわえると約2210万人にものぼるといわれている.日本人は遺伝因子として欧米人に比べてインスリンの分泌能が低いうえ(約1/2ともいわれる),環境因子として急速に進んだ食生活の欧米化や運動不足により肥満や内臓脂肪の蓄積にともなうインスリン抵抗性(インスリンが効きにくい状態)が進んだ結果,相対的なインスリンの作用不足におちいり2型糖尿病の急増にいたっていると考えられている.インスリンの主要な作用臓器として肝臓と骨格筋があげられるが,とくに骨格筋はヒトにおいて最大のグルコースの消費臓器であり,骨格筋におけるインスリン抵抗性は2型糖尿病やメタボリックシンドロームをひき起こし心筋梗塞や脳卒中といった大血管合併症や腎症や網膜症といった細小血管合併症の発症要因となっている1,2).したがって,骨格筋におけるインスリンによる糖の取り込み機構の解明や肥満によるインスリン抵抗性の発症の分子機構を明らかにすることは,糖尿病およびメタボリックシンドロームやその合併症の予防や治療において最大のテーマのひとつになっている.
骨格筋においてインスリンが正常に作用するためには,食後に膵臓のランゲルハンス島β細胞から分泌されたインスリンが骨格筋に分布する毛細血管に到達し,毛細血管が拡張して骨格筋間質に移行したインスリンが骨格筋の細胞の表面に存在するインスリン受容体に結合する必要がある3-6)(図1).以前より,骨格筋ではインスリンの移行が血液中や肝臓に比べゆっくりでありインスリン依存性の糖の取り込みも緩徐に起こることが知られている1,7,8).骨格筋細胞を用いたin vitroの実験や骨格筋間質にインスリンを直接に投与したin vivoの実験ではインスリン依存性の糖の取り込みはすみやかに起こることから,骨格筋においてインスリン依存性の糖の取り込みが緩徐な理由はインスリンの移行がゆっくりであるためと理解されている9).さらに,肥満者では血液や肝臓でのインスリンの分布に健常者と差は認められないものの,骨格筋へのインスリンの移行が低下しており,それにともなってインスリン依存性の糖の取り込みが低下していることが知られている10).実際に骨格筋間質のインスリンの濃度を測定した報告では,肥満者ではインスリンの移行速度や濃度が低下している10).こうした一連の知見は,骨格筋ではインスリンの移行が律速段階となりインスリン作用の出現が緩徐となっていること,さらに,肥満にともなう骨格筋のインスリン抵抗性には骨格筋そのものの糖の取り込みの障害にくわえ,生理的に緩徐なインスリンの移行が肥満ではさらに低下しており骨格筋における正常なインスリン依存性の糖の取り込みの障害の一因となっていることを強く示唆する.しかしこれまで,そもそも骨格筋へのインスリンの移行がどのように制御されているのか,そして,肥満ではなぜこれが障害されるのか,その分子機構は十分にわかっていなかった.今回,筆者らは,血管内皮細胞におけるインスリンシグナルが毛細血管の拡張を制御しこれにより骨格筋間質へのインスリンの移行が調節されていること,そして,肥満者では血管内皮細胞のインスリンシグナルの障害がインスリンの移行の低下の原因となっていることを明らかにした.
1.血管内皮細胞に特異的なIRS2ノックアウトマウスでは骨格筋間質へのインスリンの移行が障害されており骨格筋でのインスリン依存性の糖の取り込みが障害されていた
血管内皮細胞でのインスリン受容体の主要な基質であるIRS2(insulin receptor substrate 2)を血管内皮細胞に特異的に欠損したマウスでは,血管内皮細胞において血管拡張作用をもつNOを産生する酵素である内皮型NO合成酵素のインスリンによる活性化が約1/3~1/4に低下しており,グルコースクランプ法によりインスリン感受性を測定したところ骨格筋においてインスリンによる糖の取り込みが有意に低下していた.単離した骨格筋においてはインスリン依存性および非依存性の糖の取り込みに異常は認められなかったことから,このノックアウトマウスではインスリンの移行が障害されていることが示唆された.まず,マイクロバブルを用いて毛細血管の拡張能について野生型マウスを用いて検討したところインスリンにより有意な拡張が認められ,この拡張は内皮型NO合成酵素の阻害剤であるL-NAMEの投与により完全に阻害されることから内皮型NO合成酵素に依存性であることが確認された.そして,血管内皮細胞に特異的なIRS2ノックアウトマウスではこのインスリンによる毛細血管の拡張が有意に障害されていた.さらに,微小透析(マイクロダイアリシス)プロープを用いて骨格筋間質におけるインスリンの濃度を測定したところ,このノックアウトマウスではインスリンの移行が有意に低下していることが明らかになった.以上の結果から,血管内皮細胞に特異的なIRS2ノックアウトマウスではインスリンによる内皮型NO合成酵素の活性化が十分に起こらないため毛細血管の拡張や骨格筋間質へのインスリンの移行が障害され,骨格筋におけるインスリン依存性の糖の取り込みが低下していることが明らかになった.
血管内皮細胞に特異的なIRS2ノックアウトマウスでは全身の血管内皮細胞においてIRS2が欠損しているにもかかわらず,なぜ肝臓ではインスリン抵抗性が認められずに骨格筋においてのみインスリン抵抗性が認められたのであろうか.毛細血管はその構造から3つのタイプが存在することが知られている.骨格筋や心筋などに分布する閉鎖結合によって連続的に血管内皮細胞が連結された連続性毛細血管と,腎糸球体や内分泌腺,害分泌腺などに分布する血管内皮細胞の薄膜化した部分に大きさの一様な孔のある有窓性(小孔性)毛細血管,そして,肝臓や脾臓に分布する比較的直径が大きくアルブミンや一部の血球成分の行き来が可能な孔と断裂が認められる洞様毛細血管である.おそらく,肝臓ではインスリンはこの孔や断裂をつうじて非常にすみやかに移行することができるのに対し,連続性毛細血管が分布する骨格筋ではインスリンによる毛細血管の拡張がその移行に重要な役割をはたしているため,血管内皮細胞に特異的なIRS2ノックアウトマウスでは骨格筋においてのみインスリン抵抗性が認められたものと思われた.また,さきに述べた,インスリン移行の速度が肝臓では血液中と同じくらいすみやかであるのに対し骨格筋では緩徐であることも,このそれぞれの臓器に分布する毛細血管の構造の違いに密接に関連するものと思われる.
2.血管内皮細胞に特異的なIRS2ノックアウトマウスの骨格筋でのインスリン抵抗性はプロスタグランジンI2アナログの投与により改善した
つぎに,血管内皮細胞に特異的なIRS2ノックアウトマウスで認められた表現型をインスリンによる内皮型NO合成酵素の活性化を回復することによりレスキューすることができるかどうか検討した.プロスタグランジンI2アナログはcAMP-プロテインキナーゼA-転写因子CREBを介して血管内皮細胞において内皮型NO合成酵素の発現量を増加させることが報告されている.そこで,このノックアウトマウスにプロスタグランジンI2アナログのひとつであるベラプロストナトリウムを投与し内皮型NO合成酵素の発現量が約2倍となるようにしたところ,血管内皮細胞におけるインスリンによる内皮型NO合成酵素の活性化は野生型マウスとほぼ同じ程度にまで改善し,毛細血管の拡張能も同様に回復した.このベラプロストナトリウムによる毛細血管の拡張能の回復は内皮型NO合成酵素の阻害薬であるL-NAMEにより完全に阻害されたことから内皮型NO合成酵素を介しているものと考えられた.そして,毛細血管の拡張能の回復にともない骨格筋間質へのインスリンの移行もほぼ完全に回復し,骨格筋におけるインスリン依存性の糖の取り込みもほぼ完全に改善した.なお,ベラプロストナトリウムが骨格筋そのものの糖の取り込みに影響をあたえなかったことについては,ベラプロストナトリウムを投与したマウスの骨格筋を単離してインスリン依存性および非依存性の糖の取り込みを検討し,ベラプロストナトリウム非投与群と差が認められないことで確認した.以上の結果から,骨格筋間質へのインスリンの移行とそれにともなうインスリン依存性の骨格筋での糖の取り込みが血管内皮細胞のインスリンシグナル(インスリンによる内皮型NO合成酵素の活性化)により調節されていることが明らかになった.
3.肥満モデルマウスでは血管内皮細胞におけるIRS2の発現が低下し骨格筋間質へのインスリンの移行が障害されていた
つぎに,肥満で認められる骨格筋のインスリン抵抗性に同様な分子機構が存在するかどうかについて検討を行った.肥満モデルマウスとして8週間にわたり高脂肪食を負荷したマウスを使用した.高脂肪食負荷マウスの血管内皮細胞ではIRS1の発現が約1/2に,IRS2の発現が1/4に低下しており,それにともなってインスリンによる内皮型NO合成酵素の活性化が血管内皮細胞に特異的なIRS2ノックアウトマウスと同様に約1/3に低下していた.この高脂肪食負荷マウスの毛細血管の拡張能および骨格筋間質へのインスリンの移行を検討したところ,普通食負荷マウスに比べいずれも有意に低下していた.そして,グルコースクランプ法によりインスリン感受性を測定した結果,血管内皮細胞に特異的なIRS2ノックアウトマウスとは異なり肝臓におけるインスリン抵抗性は認められたが,骨格筋におけるインスリン依存性の糖の取り込みも有意に障害されていた.
もし,血管内皮細胞に特異的なIRS2ノックアウトマウスと同じような分子機構でこの骨格筋におけるインスリン抵抗性が生じているのであれば,同様にプロスタグランジンI2アナログであるベラプロストナトリウムによるレスキューが可能なはずである.そこで,さきほどと同様に内皮型NO合成酵素の発現量が約2倍に増加するようベラプロストナトリウムを投与したところ,インスリンによる内皮型NO合成酵素の活性化および毛細血管の拡張能はほぼ普通食負荷マウスと同じ程度にまで改善した.ベラプロストナトリウムによる毛細血管の拡張能の改善作用は内皮型NO合成酵素阻害薬であるL-NAMEにより完全に阻害されたことから内皮型NO合成酵素を介しているものと考えられた.そして,毛細血管の拡張能の改善にともない骨格筋間質へのインスリンの移行もほぼ普通食負荷マウスと同じ程度にまで回復した.グルコースクランプ法により高脂肪食負荷マウスのインスリン抵抗性を測定したところ,肝臓におけるインスリン抵抗性はまったく改善していなかったが骨格筋における糖の取り込みは有意に改善していた.しかし,その改善は毛細血管の拡張能やインスリンの移行とは異なり2/3程度にとどまっていた.これは,ベラプロストナトリウムによりインスリンの移行は改善したが肥満によってもたらされた骨格筋そのもののインスリン作用は改善しなかったためと考えられた.実際に,単離した骨格筋のインスリン依存性および非依存性の糖の取り込みを検討したところ高脂肪食負荷マウスでは普通食負荷マウスに比べ有意に低下しており,これはベラプロストナトリウムを投与してもまったく改善が認められなかった.
おわりに
以上の結果より,健常者では食後にインスリンが分泌されると内皮細胞の内皮型NO合成酵素が活性化されて毛細血管が拡張しインスリンが骨格筋間質に移行して正常に糖の取り込みが起こるのに対し,肥満者では血管内皮細胞におけるインスリン受容体基質,とくにIRS2の発現が低下しているため,食後にインスリンが分泌されてもそのシグナルは十分に伝達されずに内皮型NO合成酵素の活性化が低下し,毛細血管の拡張や骨格筋間質へのインスリンの移行が正常に起こらず骨格筋における糖の取り込みが障害されているものと考えられた(図2).そして,プロスタグランジンI2アナログであるベラプロストナトリウムはインスリンによる血管内皮細胞における内皮型NO合成酵素の活性化を改善することで,毛細血管の拡張能および骨格筋間質へのインスリンの移行を改善し骨格筋における糖の取り込みを回復することが明らかになった.これまで糖尿病の治療薬としてさまざまな分子機構をもつ薬剤が臨床で使用されているが,血管内皮細胞をターゲットとした薬剤はいうまでもなく,骨格筋へのインスリンの移行に注目した薬剤もまったく存在しないのが現状である.これまでに登場した糖尿病治療薬ではまだ十分に血糖コントロールのできていない背景の一部に,こうしたまだわれわれがアプローチできていないインスリン抵抗性の分子機構のある可能性は否定できない.今回,使用したベラプロストナトリウムは幸いにも閉塞性動脈硬化症の治療薬としてすでに臨床応用されている.今回の発見が肥満にともなうインスリン抵抗性改善薬の新たな開発につながれば幸いである.
文 献
- DeFronzo, R. A., Tobin, J. D. & Andres, R.: Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am. J. Physiol., 237, E214-E223 (1979)[PubMed]
- Bergman, R. N.: Lilly Lecture: toward physiological understanding of glucose tolerance: minimal-model approach. Diabetes, 38, 1512-1527 (1989)[PubMed]
- White, M. F. & Kahn, C. R.: The insulin signaling system. J. Biol. Chem., 269, 1-4 (1994)[PubMed]
- Petersen, K. F., Dufour, S., Befroy, D. et al.: Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med., 350, 664-671 (2004)[PubMed]
- Vincent, M. A., Clerk, L. H., Rattigan, S. et al.: Active role for the vasculature in the delivery of insulin to skeletal muscle. Clin. Exp. Pharmacol. Physiol., 32, 302-307 (2005)[PubMed]
- Long, Y. C. & Zierath, J. R.: Influence of AMP-activated protein kinase and calcineurin on metabolic networks in skeletal muscle. Am. J. Physiol. Endocrinol. Metab., 295, E545-E552 (2008)[PubMed]
- Sherwin, R. S., Kramer, K. J., Tobin, J. D. et al.: A model of the kinetics of insulin in man. J. Clin. Invest., 53, 1481-1492 (1974)[PubMed]
- Yang, Y. J., Hope, I. D., Ader, M. et al.: Insulin transport across capillaries is rate limiting for insulin action in dogs. J. Clin. Invest., 84, 1620-1628 (1989)[PubMed]
- Chiu, J. D., Richey, J. M., Harrison, L. N. et al.: Direct administration of insulin into skeletal muscle reveals that the transport of insulin across the capillary endothelium limits the time course of insulin to activate glucose disposal. Diabetes, 57, 828-835 (2008)[PubMed]
- Sjostrand, M., Gudbjornsdottir, S., Holmang, A. et al.: Delayed transcapillary transport of insulin to muscle interstitial fluid in obese subjects. Diabetes, 51, 2742-2748 (2002)[PubMed]
著者プロフィール
略歴:2001年 東京大学大学院医学系研究科 修了,2004年 東京大学医学部附属病院 客員助教を経て,2007年より東京大学システム疾患生命科学による先端医療技術開発拠点 特任准教授.
研究テーマ:モデル動物を用いた糖尿病・肥満・動脈硬化症の分子機構の解明.
窪田 哲也(Tetsuya Kubota)
国立健康・栄養研究所栄養療法プロジェクト プロジェクトリーダー.
門脇 孝(Takashi Kadowaki)
東京大学大学院医学系研究科 教授.
© 2011 窪田直人・窪田哲也・門脇 孝 Licensed under CC 表示 2.1 日本
