T細胞における内在性のRIP2による病原性Th17細胞への分化の制御
島田 賢一
(米国Cedars-Sinai Medical Center,Division of Pediatrics Infectious Disease and Immunology)
email:島田賢一
DOI: 10.7875/first.author.2018.115
T cell-intrinsic receptor interacting protein 2 regulates pathogenic T helper 17 cell differentiation.
Kenichi Shimada, Rebecca A. Porritt, Janet L. Markman, Jacqueline Gire O’Rourke, Daiko Wakita, Magali Noval Rivas, Chihiro Ogawa, Lina Kozhaya, Gislâine A. Martins, Derya Unutmaz, Robert H. Baloh, Timothy R. Crother, Shuang Chen, Moshe Arditi
Immunity, 49, 873-885.e7 (2018)
RIP2は細胞における自然免疫受容体であるNOD1およびNOD2の下流のシグナル伝達タンパク質として同定されたが,T細胞における役割については不明であった.この研究において,筆者らは,RIP2を欠損したT細胞においてインターロイキン17Aの産生が促進され,クラミジア肺炎,動脈硬化,実験的自己免疫性脳脊髄炎が増悪することを明らかにした.また,RIP2の欠損により非病原性Th17細胞への分化は抑制されるのに対し病原性Th17細胞への分化が促進され,それには転写因子RORαおよびインターロイキン1のシグナルを必要としたが,NOD1およびNOD2には非依存性であった.さらには,T細胞におけるRIP2のCARDドメインの過剰な発現は病原性Th17細胞の分化を有意に減弱させた.この研究により,T細胞においてRIP2が病原性Th17細胞および非病原性Th17細胞への分化の恒常性を制御することが明らかにされた.
インターロイキン17Aを産生するTh17細胞は自己免疫疾患,感染症,大腸炎,がん,皮膚疾患など,さまざまな病態において重要な役割を担うという認識が定着した1).TGFβおよびインターロイキン6の共存在下において通常の非病原性Th17細胞への分化が誘導され,RORγt,IRF4,BATFなどの転写因子がこれらの細胞のプログラミングに必要であると報告された2).その一方で,インターロイキン1β,インターロイキン6,インターロイキン23の共刺激により分化したTh17細胞はより炎症性の高いことから病原性Th17細胞として区別され,さまざまな研究分野において注目されてきた3).
RIP2は細胞における自然免疫受容体であるNOD1およびNOD2による細菌の細胞壁に含まれるペプチドグリカンの認識を担うシグナル伝達タンパク質として同定され4,5),マクロファージ,樹状細胞,好中球といったおもにミエロイド系細胞において重要とされていたが,T細胞における役割は不明のままであった.さきの報告において,RIP2はTh1細胞およびTh2細胞の応答に重要であるとされたものの6,7),のちの報告によりこれは否定された8).しかしながら,Th17細胞への関与についてはいまだ明らかにされていない.この研究において,筆者らは,RIP2を欠損したCD4陽性T細胞はTh17細胞への分化が促進されていることを見い出し,RIP2はT細胞においてTh17細胞への分化を負に制御することを明らかにした.
以前に,筆者らは,RIP2ノックアウトマウスは肺へのクラミジアの感染において感染防御における応答能がいちじるしく低下し慢性炎症をひき起こすことを報告した9).これは,肺においてマクロファージの活性化能が低下しクラミジアの排除が遅延するためであった.この肺へのクラミジアの感染において,RIP2ノックアウトマウスはインターロイキン17Aの産生を促進することが認められた.ほかの細菌の肺への感染モデルにおいてγδT細胞やナチュラルキラー細胞がインターロイキン17Aの産生にかかわるとの報告のあったことから,肺へのクラミジアの感染ののちインターロイキン17Aを産生する細胞を経時的に調べたところ,感染の後期においてCD4陽性T細胞からのインターロイキン17Aの産生がRIP2ノックアウトマウスにおいて有意に増加することが判明した.クラミジアが感染した肺の流入領域のリンパ節細胞を再刺激すると,RIP2ノックアウトマウスにおいてインターロイキン17Aの産生は顕著に増加し,逆に,インターフェロンγの産生は減少した.このことから,RIP2の欠損はTh17細胞への分化に強くかかわることが示された.しかし,RIP2の欠損がT細胞において直接的に関与するのか,それとも,環境作用としてT細胞の分化に関与しインターロイキン17Aの産生を促進したのかは不明であった.そこで,抗原提示細胞である樹状細胞にクラミジアの抗原を取り込ませ流入領域のリンパ節細胞を再刺激したところ,同様に,RIP2を欠損したT細胞においてTh17細胞への分化の顕著な促進が認められた.このことから,T細胞においてRIP2が直接的にTh17細胞への分化に関与する可能性が示された.さらに,この現象が生体においてもみられるのか確認するため,RAG1ノックアウトマウスにRIP2を欠損したCD4陽性T細胞を移入し,肺へクラミジアを感染させた.さきと同様に,流入領域のリンパ節細胞を再刺激するとRIP2を欠損したCD4陽性T細胞を移入したマウスにおいてインターロイキン17Aの産生は顕著に増加し,インターフェロンγの産生は顕著に減少した.これらのことから,T細胞におけるRIP2の欠損によりTh17細胞への分化が促進されることが示され,RIP2はTh17細胞への分化を負に制御することが明らかにされた.
RIP2ノックアウトマウスは肺へのクラミジアの感染においてTh17細胞への分化が顕著に促進されたが,それはRIP2のT細胞における内在的な役割によるものであった.しかしながら,これらがT細胞の前プログラミングによるものなのか,T細胞の再刺激のときに起こる現象であるのかは不明であった.そこで,それぞれのナイーブCD4陽性T細胞を,TGFβおよびインターロイキン6により分化される非病原性Th17細胞,および,インターロイキン1β,インターロイキン6,インターロイキン23により分化される病原性Th17細胞へと分化させることを試みた.すると,RIP2を欠損したT細胞は,非病原性Th17細胞への分化は減弱し病原性Th17細胞への分化は亢進した.同様のTh17細胞への分化の実験をNOD2を欠損したナイーブCD4陽性T細胞およびNOD1とNOD2を二重欠損したナイーブCD4陽性T細胞において実施したが,RIP2を欠損したナイーブCD4陽性T細胞のようなTh17細胞の分化の変化は認められなかった.このことから,RIP2はNOD1およびNOD2とは独立してTh17細胞の分化を制御することが判明した.
RIP2ノックアウトマウスは肺へのクラミジアの感染において感染防御における応答能がいちじるしく低下し慢性炎症をひき起こすことが見い出されたが9),これがTh17細胞によるインターロイキン17Aの産生によるものなのか,また,インターロイキン17Aは肺へのクラミジアに感染においてどのような役割を担うのかは不明であった.インターロイキン17Aはさまざまな細菌や菌類に対する感染防御に重要な役割を担うことは報告されていたが,おもに細胞の外に存在する細菌に対しての役割であり,クラミジアなどの細胞の内部に寄生した細菌に対するインターロイキン17Aの役割はいまだ明らかではない.そこで,インターロイキン17Aノックアウトマウスにクラミジアを感染させたところ,クラミジアの除去能は野生型のマウスと同等であったのにもかかわらず,クラミジアによる肺炎から早期に回復した.クラミジアに感染した初期の好中球の肺への浸潤に差はなかったが,野生型マウスのほうが好中球が肺に持続しつづける傾向が認められ,それにともないCD4陽性T細胞の持続が認められた.これらのことから,インターロイキン17Aはクラミジアの排除にはかかわらないが,クラミジアの感染によりひき起こされる炎症の慢性化に関与することが示された.そこで,RIP2ノックアウトマウスにおけるクラミジアの感染による慢性肺炎がインターロイキン17Aによるものであるかどうかを明らかにするため,RIP2とインターロイキン17Aのダブルノックアウトマウスを作製したところ,RIP2ノックアウトマウスと同様に,感染の初期におけるクラミジアの除去に遅延が認められたが,感染の後期における慢性肺炎は野生型のマウスと同等に回復した.以上のことから,RIP2の欠損により増悪したクラミジアの感染による慢性肺炎はインターロイキン17Aによるものであることが明らかにされた.
RIP2の欠損により分化が亢進した病原性Th17細胞による病態の悪性化は,細菌感染症だけでなくほかの疾患の進展にも寄与すると考えられた1).そこで,高脂肪食によりひき起されるマウスの実験的な動脈硬化のモデルを用いた.RIP2を欠損した骨髄細胞,RIP2およびインターロイキン17Aを欠損した骨髄細胞,野生型の骨髄細胞を,放射線を照射したLDL受容体のノックアウトマウスに移植し,高脂肪食をあたえて動脈硬化の進行について評価したところ,RIP2を欠損した骨髄を移植したマウスにおいては野生型の骨髄を移殖したマウスに比べ動脈硬化が顕著に進行したのに対し,RIP2およびインターロイキン17Aを欠損した骨髄を移殖したマウスには動脈硬化の促進は認められなかった.しかしながら,用いた実験系は移殖した骨髄細胞がすべての造血細胞と置き換わるモデルであり,T細胞以外の造血細胞のRIP2が寄与した可能性も考えらえた.そこで,RIP2を欠損したCD4陽性T細胞をRAG1ノックアウトマウスに直接的に移入し,そののち,アデノウイルスベクターを用いてPCSK9遺伝子を強制的に発現させ,高脂肪食をあたえて動脈硬化について評価した.PCSKは肝臓においてLDL受容体の発現の低下をひき起こしLDL受容体のノックアウトマウスと同様に動脈硬化を促進させる,近年,新たに提唱された実験モデルである.その結果,骨髄を移植したLDL受容体のノックアウトマウスほど進行した動脈硬化はみられなかったが,野生型のCD4陽性T細胞を移入したマウスに比べ,RIP2を欠損したCD4陽性T細胞を移入したマウスは動脈硬化が顕著に促進された.これに対し,RIP2およびインターロイキン17Aを欠損したCD4陽性T細胞を移入したマウスには動脈硬化の促進は認められなかった.以上のことから,T細胞に由来するRIP2の欠損がインターロイキン17Aに依存的に動脈硬化を進行させることが明らかにされた.
これらのことから,T細胞に由来するRIP2の欠損がインターロイキン17Aに依存的に病態を悪性化させたことは証明されたが,これを病原性のTh17細胞によるものと断定することは困難であった.T細胞が動脈硬化を進行させることは既存の理解にあったが,T細胞の抗原性は不明であったからである.そこで,自己抗原に対する免疫反応により病態が進行し病原性のTh17細胞が重要であると認識されている実験的自己免疫性脳脊髄膜炎モデルを用いて,T細胞に由来するRIP2の病原性について再評価した.RIP2ノックアウトマウスを用いた実験的自己免疫性脳脊髄膜炎は2011年に報告されており10),RIP2ノックアウトマウス,Nod1ノックアウトマウス,Nod2ノックアウトマウスともに実験的自己免疫性脳脊髄膜炎に対し抵抗性を示すという結果であった.しかしながら,実験的自己免疫性脳脊髄膜炎モデルそのものはミエリンに由来するMOGペプチドをCFAアジュバントにより免疫することによりひき起こすものであり,CFAアジュバントそれ自体に結核菌の細胞壁の成分が含まれる.ミエロイド系細胞のNOD1-RIP2シグナル伝達経路およびNOD2-RIP2シグナル伝達経路それ自体,本来は結核菌の細胞壁を認識するものであることから,病態にRIP2がかかわるというよりは免疫の成立に抵抗を示したことにほかならず,RIP2が自己免疫性脳脊髄膜炎の進行に関与するかどうかは評価されていないと考えられた10).そこで,RIP2を欠損したCD4陽性T細胞を移入したRAG1ノックアウトマウスをMOGペプチドおよびCFAアジュバントにより免疫して実験的自己免疫性脳脊髄膜炎をひき起こした.その結果,以前の報告とは異なり,RIP2を欠損したCD4陽性T細胞を移入したマウスは野生型のCD4陽性T細胞を移入したマウスよりもはるかに自己免疫性脳脊髄膜炎の悪性化が認められ,それにともない致死率も上昇した.これらの結果から,T細胞におけるRIP2の欠損により病原性Th17細胞の分化が誘導され,実際に生体において病態を悪性化させることが明らかにされた.
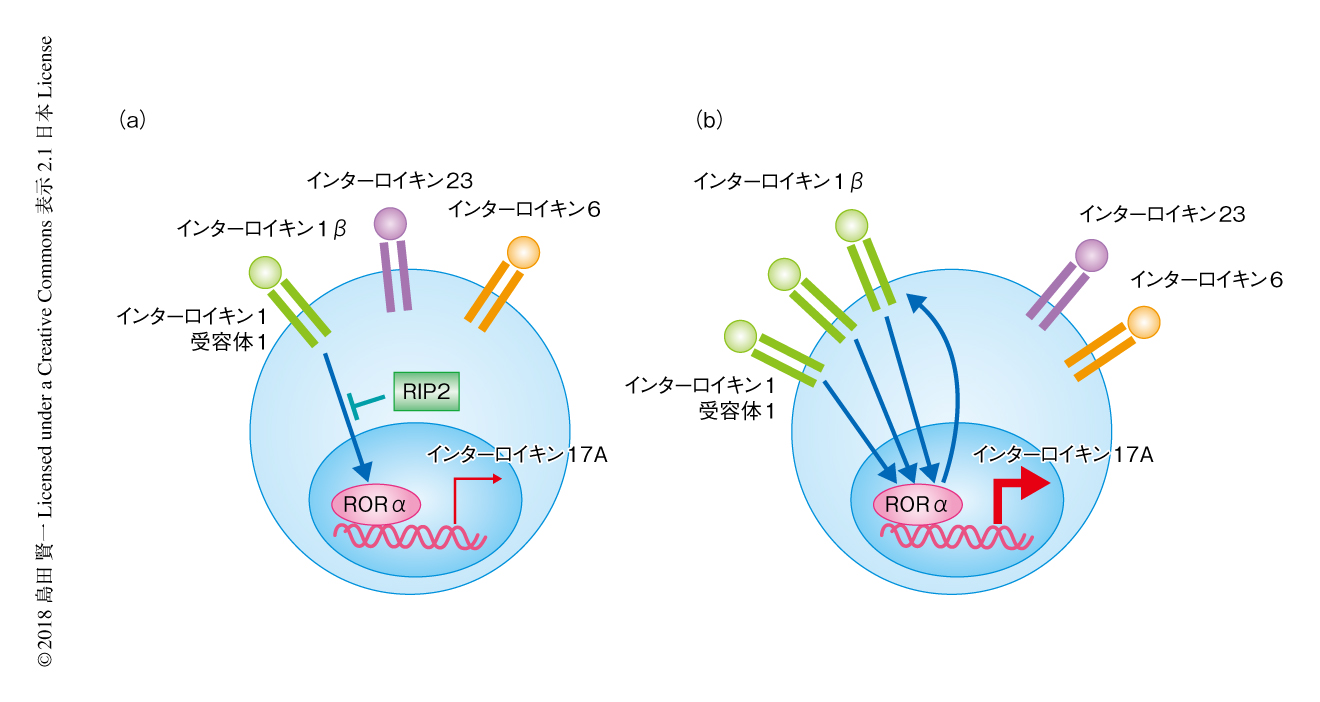
RORγt,IRF4,BATFなどの転写因子によりTh17細胞への分化が誘導されることが報告されていた.しかしながら,RIP2を欠損したT細胞から分化した病原性Th17細胞におけるこれらの転写因子の発現は野生型のT細胞と同等であったのに対し,RORαの発現はmRNAレベルおよびタンパク質レベルで上昇していた.そこで,siRNAおよびshRNAを用いてRORαをノックダウンしたところ,RIP2を欠損したT細胞に認められたインターロイキン17Aの産生の増加が消失した.RIP2を欠損したT細胞から分化した病原性Th17細胞においてはインターロイキン1受容体1のmRNAレベルでの発現が顕著に上昇していたが,RORαのノックダウンによりこの発現の上昇も消失した.このことから,RIP2を欠損したT細胞の病原性Th17細胞への分化は転写因子RORαおよびインターロイキン1βからのシグナルに依存することが明らかにされた(図1).
これまで,RIP2の欠損における病原性Th17細胞への分化の促進を観察してきたが,このことから,RIP2は病原性Th17細胞への分化を負に制御することが示唆された.そこで,実際の病原性Th17細胞への分化におけるRIP2の発現について調べたところ,mRNAレベルの発現はT細胞受容体への刺激により上昇したのに対し,病原性Th17細胞が分化する条件のT細胞受容体への刺激およびサイトカインの共刺激では低下した.これらは,さきの発見と同様に,RIP2の発現のレベルがTh17細胞への分化に関与することを示していた.これらRIP2の発現の低下が病態の進行しているTh17細胞においても起こるのかどうか検討した.インターフェロンγおよびインターロイキン17Aを蛍光タンパク質により標識したレポーターマウスにクラミジアを感染させ,インターフェロンγを産生するT細胞とインターロイキン17Aを産生するT細胞とをセルソーターにより単離してRIP2の発現について調べたところ,インターフェロンγを産生するT細胞と比べインターロイキン17Aを産生するT細胞はRIP2の発現が低下していた.このことから,RIP2のT細胞における発現の上下は病原性Th17細胞の分化と負に連動することが証明された.そこで,T細胞においてRIP2を強制的に発現させたらどう変化するのか検討した.RIP2をレトロウイルスベクターを用いて野生型のCD4陽性T細胞において強制発現させると病原性Th17細胞への分化がいちじるしく減少した.
RIP2にはキナーゼ活性部位があり,RIP2のキナーゼ活性がミエロイド系細胞におけるNod1シグナル伝達経路およびNod2シグナル伝達経路において重要であるとされていた.そこで,RIP2のキナーゼ活性部位に点変異を導入し同様に野生型のCD4陽性T細胞に強制発現させたが,やはり病原性Th17細胞への分化の減少がみられた.RIP2のキナーゼ活性部位はT細胞から病原性Th17細胞への分化には重要ではなかった.そこで,RIP2のCARDドメインを欠損した変異体をT細胞に強制発現させると,病原性Th17細胞への分化の減少が消失した.さらに,RIP2のCARDドメインを単独でT細胞に強制発現させたところ,病原性Th17細胞への分化はいちじるしく減少した.このことから,RIP2のCARDドメインが病原性Th17細胞への分化を制御することが明らかにされた.合成したRIP2のCARDドメインに細胞透過性ペプチドを付加し,遺伝子ではなくタンパク質として直接的にT細胞に導入したところ,遺伝子の導入と同じくタンパク質として導入しても病原性Th17細胞への分化が制御され,治療薬としての可能性のあることが見い出された.
2000年代初頭に自然免疫においてNOD1シグナル伝達経路およびNOD2シグナル伝達経路を構成するタンパク質としての役割が確立されて以来,RIP2はあくまでミエロイド系細胞および抗原提示能をもつ一部の上皮細胞や血管内皮細胞において役割を担うという認識にとどまっていた.この研究は,RIP2のT細胞における内在的な役割をはじめて立証したといえよう.また,これまではNOD-RIP2シグナル伝達経路は細菌の感染による細菌の細胞壁に含まれるペプチドグリカンの認識にはたらくという理解であったが,ペプチドグリカンのかかわらないウイルスの感染系においても重要であったり,動脈硬化や心筋虚血においてはこれを抑制していたりと,細菌の感染のほかの病態においてもその関与が示唆されるような報告が増えたことは興味深い.
RIP2がどのようにRORαを制御しているのかはこれからの課題であるが,Rip2遺伝子の発現制御部位にはRORαが転写因子として認識しうる配列がヒトおよびマウスにおいて存在する.このことから,RORαがRip2遺伝子の転写を制御するフィードバック機構の存在することが推察される.
RIP2のCARDドメインがどのような機構によりTh17細胞への分化を抑制するのかも今後の課題であるが,RIP2のCARDドメインのT細胞への選択的な輸送が可能になれば,現在,検討されているヒト抗インターロイキン17抗体を用いた乾癬などを対象にした臨床応用よりもすぐれた効果が期待できるかもしれない.非病原性Th17細胞に由来するインターロイキン17Aは小腸の炎症にはむしろ抑制的にはたらくことが報告され,ヒト抗インターロイキン17抗体は皮膚疾患や脊椎炎などを治療する代わりに小腸の炎症を誘発してしまう可能性がありこの懸念が先行しているが,RIP2のCARDドメインは病原性Th17細胞のみを阻害するという点ではすぐれているからである.
略歴:2006年 北里大学大学院基礎生命科学研究科博士課程 修了,同年 米国Cedars-Sinai Medical CenterにてPost Doctoral Fellow,2009年 同Research Scientist,2012年 同Assistant Professorを経て,2018年より同Associate Professor.
研究テーマ:感染免疫,アレルギー,動脈硬化,急性肺傷害,川崎病.
© 2018 島田 賢一 Licensed under CC 表示 2.1 日本
(米国Cedars-Sinai Medical Center,Division of Pediatrics Infectious Disease and Immunology)
email:島田賢一
DOI: 10.7875/first.author.2018.115
T cell-intrinsic receptor interacting protein 2 regulates pathogenic T helper 17 cell differentiation.
Kenichi Shimada, Rebecca A. Porritt, Janet L. Markman, Jacqueline Gire O’Rourke, Daiko Wakita, Magali Noval Rivas, Chihiro Ogawa, Lina Kozhaya, Gislâine A. Martins, Derya Unutmaz, Robert H. Baloh, Timothy R. Crother, Shuang Chen, Moshe Arditi
Immunity, 49, 873-885.e7 (2018)
要 約
RIP2は細胞における自然免疫受容体であるNOD1およびNOD2の下流のシグナル伝達タンパク質として同定されたが,T細胞における役割については不明であった.この研究において,筆者らは,RIP2を欠損したT細胞においてインターロイキン17Aの産生が促進され,クラミジア肺炎,動脈硬化,実験的自己免疫性脳脊髄炎が増悪することを明らかにした.また,RIP2の欠損により非病原性Th17細胞への分化は抑制されるのに対し病原性Th17細胞への分化が促進され,それには転写因子RORαおよびインターロイキン1のシグナルを必要としたが,NOD1およびNOD2には非依存性であった.さらには,T細胞におけるRIP2のCARDドメインの過剰な発現は病原性Th17細胞の分化を有意に減弱させた.この研究により,T細胞においてRIP2が病原性Th17細胞および非病原性Th17細胞への分化の恒常性を制御することが明らかにされた.
はじめに
インターロイキン17Aを産生するTh17細胞は自己免疫疾患,感染症,大腸炎,がん,皮膚疾患など,さまざまな病態において重要な役割を担うという認識が定着した1).TGFβおよびインターロイキン6の共存在下において通常の非病原性Th17細胞への分化が誘導され,RORγt,IRF4,BATFなどの転写因子がこれらの細胞のプログラミングに必要であると報告された2).その一方で,インターロイキン1β,インターロイキン6,インターロイキン23の共刺激により分化したTh17細胞はより炎症性の高いことから病原性Th17細胞として区別され,さまざまな研究分野において注目されてきた3).
RIP2は細胞における自然免疫受容体であるNOD1およびNOD2による細菌の細胞壁に含まれるペプチドグリカンの認識を担うシグナル伝達タンパク質として同定され4,5),マクロファージ,樹状細胞,好中球といったおもにミエロイド系細胞において重要とされていたが,T細胞における役割は不明のままであった.さきの報告において,RIP2はTh1細胞およびTh2細胞の応答に重要であるとされたものの6,7),のちの報告によりこれは否定された8).しかしながら,Th17細胞への関与についてはいまだ明らかにされていない.この研究において,筆者らは,RIP2を欠損したCD4陽性T細胞はTh17細胞への分化が促進されていることを見い出し,RIP2はT細胞においてTh17細胞への分化を負に制御することを明らかにした.
1.T細胞におけるRIP2の欠損によりインターロイキン17Aの産生が促進される
以前に,筆者らは,RIP2ノックアウトマウスは肺へのクラミジアの感染において感染防御における応答能がいちじるしく低下し慢性炎症をひき起こすことを報告した9).これは,肺においてマクロファージの活性化能が低下しクラミジアの排除が遅延するためであった.この肺へのクラミジアの感染において,RIP2ノックアウトマウスはインターロイキン17Aの産生を促進することが認められた.ほかの細菌の肺への感染モデルにおいてγδT細胞やナチュラルキラー細胞がインターロイキン17Aの産生にかかわるとの報告のあったことから,肺へのクラミジアの感染ののちインターロイキン17Aを産生する細胞を経時的に調べたところ,感染の後期においてCD4陽性T細胞からのインターロイキン17Aの産生がRIP2ノックアウトマウスにおいて有意に増加することが判明した.クラミジアが感染した肺の流入領域のリンパ節細胞を再刺激すると,RIP2ノックアウトマウスにおいてインターロイキン17Aの産生は顕著に増加し,逆に,インターフェロンγの産生は減少した.このことから,RIP2の欠損はTh17細胞への分化に強くかかわることが示された.しかし,RIP2の欠損がT細胞において直接的に関与するのか,それとも,環境作用としてT細胞の分化に関与しインターロイキン17Aの産生を促進したのかは不明であった.そこで,抗原提示細胞である樹状細胞にクラミジアの抗原を取り込ませ流入領域のリンパ節細胞を再刺激したところ,同様に,RIP2を欠損したT細胞においてTh17細胞への分化の顕著な促進が認められた.このことから,T細胞においてRIP2が直接的にTh17細胞への分化に関与する可能性が示された.さらに,この現象が生体においてもみられるのか確認するため,RAG1ノックアウトマウスにRIP2を欠損したCD4陽性T細胞を移入し,肺へクラミジアを感染させた.さきと同様に,流入領域のリンパ節細胞を再刺激するとRIP2を欠損したCD4陽性T細胞を移入したマウスにおいてインターロイキン17Aの産生は顕著に増加し,インターフェロンγの産生は顕著に減少した.これらのことから,T細胞におけるRIP2の欠損によりTh17細胞への分化が促進されることが示され,RIP2はTh17細胞への分化を負に制御することが明らかにされた.
2.RIP2を欠損したナイーブCD4陽性T細胞は病原性Th17細胞への分化が促進される
RIP2ノックアウトマウスは肺へのクラミジアの感染においてTh17細胞への分化が顕著に促進されたが,それはRIP2のT細胞における内在的な役割によるものであった.しかしながら,これらがT細胞の前プログラミングによるものなのか,T細胞の再刺激のときに起こる現象であるのかは不明であった.そこで,それぞれのナイーブCD4陽性T細胞を,TGFβおよびインターロイキン6により分化される非病原性Th17細胞,および,インターロイキン1β,インターロイキン6,インターロイキン23により分化される病原性Th17細胞へと分化させることを試みた.すると,RIP2を欠損したT細胞は,非病原性Th17細胞への分化は減弱し病原性Th17細胞への分化は亢進した.同様のTh17細胞への分化の実験をNOD2を欠損したナイーブCD4陽性T細胞およびNOD1とNOD2を二重欠損したナイーブCD4陽性T細胞において実施したが,RIP2を欠損したナイーブCD4陽性T細胞のようなTh17細胞の分化の変化は認められなかった.このことから,RIP2はNOD1およびNOD2とは独立してTh17細胞の分化を制御することが判明した.
3.RIP2の欠損により増加したインターロイキン17Aによりクラミジアによる肺炎は慢性化する
RIP2ノックアウトマウスは肺へのクラミジアの感染において感染防御における応答能がいちじるしく低下し慢性炎症をひき起こすことが見い出されたが9),これがTh17細胞によるインターロイキン17Aの産生によるものなのか,また,インターロイキン17Aは肺へのクラミジアに感染においてどのような役割を担うのかは不明であった.インターロイキン17Aはさまざまな細菌や菌類に対する感染防御に重要な役割を担うことは報告されていたが,おもに細胞の外に存在する細菌に対しての役割であり,クラミジアなどの細胞の内部に寄生した細菌に対するインターロイキン17Aの役割はいまだ明らかではない.そこで,インターロイキン17Aノックアウトマウスにクラミジアを感染させたところ,クラミジアの除去能は野生型のマウスと同等であったのにもかかわらず,クラミジアによる肺炎から早期に回復した.クラミジアに感染した初期の好中球の肺への浸潤に差はなかったが,野生型マウスのほうが好中球が肺に持続しつづける傾向が認められ,それにともないCD4陽性T細胞の持続が認められた.これらのことから,インターロイキン17Aはクラミジアの排除にはかかわらないが,クラミジアの感染によりひき起こされる炎症の慢性化に関与することが示された.そこで,RIP2ノックアウトマウスにおけるクラミジアの感染による慢性肺炎がインターロイキン17Aによるものであるかどうかを明らかにするため,RIP2とインターロイキン17Aのダブルノックアウトマウスを作製したところ,RIP2ノックアウトマウスと同様に,感染の初期におけるクラミジアの除去に遅延が認められたが,感染の後期における慢性肺炎は野生型のマウスと同等に回復した.以上のことから,RIP2の欠損により増悪したクラミジアの感染による慢性肺炎はインターロイキン17Aによるものであることが明らかにされた.
4.RIP2を欠損したT細胞により増加したインターロイキン17Aは動脈硬化症や実験的自己免疫性脳脊髄膜炎を増悪させる
RIP2の欠損により分化が亢進した病原性Th17細胞による病態の悪性化は,細菌感染症だけでなくほかの疾患の進展にも寄与すると考えられた1).そこで,高脂肪食によりひき起されるマウスの実験的な動脈硬化のモデルを用いた.RIP2を欠損した骨髄細胞,RIP2およびインターロイキン17Aを欠損した骨髄細胞,野生型の骨髄細胞を,放射線を照射したLDL受容体のノックアウトマウスに移植し,高脂肪食をあたえて動脈硬化の進行について評価したところ,RIP2を欠損した骨髄を移植したマウスにおいては野生型の骨髄を移殖したマウスに比べ動脈硬化が顕著に進行したのに対し,RIP2およびインターロイキン17Aを欠損した骨髄を移殖したマウスには動脈硬化の促進は認められなかった.しかしながら,用いた実験系は移殖した骨髄細胞がすべての造血細胞と置き換わるモデルであり,T細胞以外の造血細胞のRIP2が寄与した可能性も考えらえた.そこで,RIP2を欠損したCD4陽性T細胞をRAG1ノックアウトマウスに直接的に移入し,そののち,アデノウイルスベクターを用いてPCSK9遺伝子を強制的に発現させ,高脂肪食をあたえて動脈硬化について評価した.PCSKは肝臓においてLDL受容体の発現の低下をひき起こしLDL受容体のノックアウトマウスと同様に動脈硬化を促進させる,近年,新たに提唱された実験モデルである.その結果,骨髄を移植したLDL受容体のノックアウトマウスほど進行した動脈硬化はみられなかったが,野生型のCD4陽性T細胞を移入したマウスに比べ,RIP2を欠損したCD4陽性T細胞を移入したマウスは動脈硬化が顕著に促進された.これに対し,RIP2およびインターロイキン17Aを欠損したCD4陽性T細胞を移入したマウスには動脈硬化の促進は認められなかった.以上のことから,T細胞に由来するRIP2の欠損がインターロイキン17Aに依存的に動脈硬化を進行させることが明らかにされた.
これらのことから,T細胞に由来するRIP2の欠損がインターロイキン17Aに依存的に病態を悪性化させたことは証明されたが,これを病原性のTh17細胞によるものと断定することは困難であった.T細胞が動脈硬化を進行させることは既存の理解にあったが,T細胞の抗原性は不明であったからである.そこで,自己抗原に対する免疫反応により病態が進行し病原性のTh17細胞が重要であると認識されている実験的自己免疫性脳脊髄膜炎モデルを用いて,T細胞に由来するRIP2の病原性について再評価した.RIP2ノックアウトマウスを用いた実験的自己免疫性脳脊髄膜炎は2011年に報告されており10),RIP2ノックアウトマウス,Nod1ノックアウトマウス,Nod2ノックアウトマウスともに実験的自己免疫性脳脊髄膜炎に対し抵抗性を示すという結果であった.しかしながら,実験的自己免疫性脳脊髄膜炎モデルそのものはミエリンに由来するMOGペプチドをCFAアジュバントにより免疫することによりひき起こすものであり,CFAアジュバントそれ自体に結核菌の細胞壁の成分が含まれる.ミエロイド系細胞のNOD1-RIP2シグナル伝達経路およびNOD2-RIP2シグナル伝達経路それ自体,本来は結核菌の細胞壁を認識するものであることから,病態にRIP2がかかわるというよりは免疫の成立に抵抗を示したことにほかならず,RIP2が自己免疫性脳脊髄膜炎の進行に関与するかどうかは評価されていないと考えられた10).そこで,RIP2を欠損したCD4陽性T細胞を移入したRAG1ノックアウトマウスをMOGペプチドおよびCFAアジュバントにより免疫して実験的自己免疫性脳脊髄膜炎をひき起こした.その結果,以前の報告とは異なり,RIP2を欠損したCD4陽性T細胞を移入したマウスは野生型のCD4陽性T細胞を移入したマウスよりもはるかに自己免疫性脳脊髄膜炎の悪性化が認められ,それにともない致死率も上昇した.これらの結果から,T細胞におけるRIP2の欠損により病原性Th17細胞の分化が誘導され,実際に生体において病態を悪性化させることが明らかにされた.
5.転写因子RORαおよびインターロイキン1βはRIP2を欠損したT細胞においてインターロイキン17Aの産生を促進する
RORγt,IRF4,BATFなどの転写因子によりTh17細胞への分化が誘導されることが報告されていた.しかしながら,RIP2を欠損したT細胞から分化した病原性Th17細胞におけるこれらの転写因子の発現は野生型のT細胞と同等であったのに対し,RORαの発現はmRNAレベルおよびタンパク質レベルで上昇していた.そこで,siRNAおよびshRNAを用いてRORαをノックダウンしたところ,RIP2を欠損したT細胞に認められたインターロイキン17Aの産生の増加が消失した.RIP2を欠損したT細胞から分化した病原性Th17細胞においてはインターロイキン1受容体1のmRNAレベルでの発現が顕著に上昇していたが,RORαのノックダウンによりこの発現の上昇も消失した.このことから,RIP2を欠損したT細胞の病原性Th17細胞への分化は転写因子RORαおよびインターロイキン1βからのシグナルに依存することが明らかにされた(図1).
6.RIP2のCARDドメインによる病原性Th17細胞への分化の抑制
これまで,RIP2の欠損における病原性Th17細胞への分化の促進を観察してきたが,このことから,RIP2は病原性Th17細胞への分化を負に制御することが示唆された.そこで,実際の病原性Th17細胞への分化におけるRIP2の発現について調べたところ,mRNAレベルの発現はT細胞受容体への刺激により上昇したのに対し,病原性Th17細胞が分化する条件のT細胞受容体への刺激およびサイトカインの共刺激では低下した.これらは,さきの発見と同様に,RIP2の発現のレベルがTh17細胞への分化に関与することを示していた.これらRIP2の発現の低下が病態の進行しているTh17細胞においても起こるのかどうか検討した.インターフェロンγおよびインターロイキン17Aを蛍光タンパク質により標識したレポーターマウスにクラミジアを感染させ,インターフェロンγを産生するT細胞とインターロイキン17Aを産生するT細胞とをセルソーターにより単離してRIP2の発現について調べたところ,インターフェロンγを産生するT細胞と比べインターロイキン17Aを産生するT細胞はRIP2の発現が低下していた.このことから,RIP2のT細胞における発現の上下は病原性Th17細胞の分化と負に連動することが証明された.そこで,T細胞においてRIP2を強制的に発現させたらどう変化するのか検討した.RIP2をレトロウイルスベクターを用いて野生型のCD4陽性T細胞において強制発現させると病原性Th17細胞への分化がいちじるしく減少した.
RIP2にはキナーゼ活性部位があり,RIP2のキナーゼ活性がミエロイド系細胞におけるNod1シグナル伝達経路およびNod2シグナル伝達経路において重要であるとされていた.そこで,RIP2のキナーゼ活性部位に点変異を導入し同様に野生型のCD4陽性T細胞に強制発現させたが,やはり病原性Th17細胞への分化の減少がみられた.RIP2のキナーゼ活性部位はT細胞から病原性Th17細胞への分化には重要ではなかった.そこで,RIP2のCARDドメインを欠損した変異体をT細胞に強制発現させると,病原性Th17細胞への分化の減少が消失した.さらに,RIP2のCARDドメインを単独でT細胞に強制発現させたところ,病原性Th17細胞への分化はいちじるしく減少した.このことから,RIP2のCARDドメインが病原性Th17細胞への分化を制御することが明らかにされた.合成したRIP2のCARDドメインに細胞透過性ペプチドを付加し,遺伝子ではなくタンパク質として直接的にT細胞に導入したところ,遺伝子の導入と同じくタンパク質として導入しても病原性Th17細胞への分化が制御され,治療薬としての可能性のあることが見い出された.
おわりに
2000年代初頭に自然免疫においてNOD1シグナル伝達経路およびNOD2シグナル伝達経路を構成するタンパク質としての役割が確立されて以来,RIP2はあくまでミエロイド系細胞および抗原提示能をもつ一部の上皮細胞や血管内皮細胞において役割を担うという認識にとどまっていた.この研究は,RIP2のT細胞における内在的な役割をはじめて立証したといえよう.また,これまではNOD-RIP2シグナル伝達経路は細菌の感染による細菌の細胞壁に含まれるペプチドグリカンの認識にはたらくという理解であったが,ペプチドグリカンのかかわらないウイルスの感染系においても重要であったり,動脈硬化や心筋虚血においてはこれを抑制していたりと,細菌の感染のほかの病態においてもその関与が示唆されるような報告が増えたことは興味深い.
RIP2がどのようにRORαを制御しているのかはこれからの課題であるが,Rip2遺伝子の発現制御部位にはRORαが転写因子として認識しうる配列がヒトおよびマウスにおいて存在する.このことから,RORαがRip2遺伝子の転写を制御するフィードバック機構の存在することが推察される.
RIP2のCARDドメインがどのような機構によりTh17細胞への分化を抑制するのかも今後の課題であるが,RIP2のCARDドメインのT細胞への選択的な輸送が可能になれば,現在,検討されているヒト抗インターロイキン17抗体を用いた乾癬などを対象にした臨床応用よりもすぐれた効果が期待できるかもしれない.非病原性Th17細胞に由来するインターロイキン17Aは小腸の炎症にはむしろ抑制的にはたらくことが報告され,ヒト抗インターロイキン17抗体は皮膚疾患や脊椎炎などを治療する代わりに小腸の炎症を誘発してしまう可能性がありこの懸念が先行しているが,RIP2のCARDドメインは病原性Th17細胞のみを阻害するという点ではすぐれているからである.
文 献
- Patel, D. D. & Kuchroo, V. K.: Th17 cell pathway in human immunity: lessons from genetics and therapeutic interventions. Immunity, 43, 1040-1051 (2015)[PubMed]
- Veldhoen, M., Hocking, R. J., Atkins, C. J. et al.: TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity, 24, 179-189 (2006)[PubMed]
- Ghoreschi, K., Laurence, A., Yang, X. P. et al.: Generation of pathogenic TH17 cells in the absence of TGF-β signalling. Nature, 467, 967-971 (2010)[PubMed]
- Girardin, S. E., Travassos, L. H., Herve, M. et al.: Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J. Biol. Chem., 24, 41702-41708 (2003)[PubMed]
- Ogura, Y., Inohara, N., Benito, A. et al.: Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-κB. J. Biol. Chem., 276, 4812-4818 (2001)[PubMed]
- Chin, A. I., Dempsey, P. W., Bruhn, K. et al.: Involvement of receptor-interacting protein 2 in innate and adaptive immune responses. Nature, 416, 190-194 (2002)[PubMed]
- Kobayashi, K., Inohara, N., Hernandez, L. D. et al.: RICK/Rip2/CARDIAK mediates signaling for receptors of the innate and adaptive immune systems. Nature, 416, 194-199 (2002)[PubMed]
- Hall, H. T., Wilhelm, M. T., Saibil, S. D. et al.: RIP2 contributes to Nod signaling but is not essential for T cell proliferation, T helper differentiation or TLR responses. Eur. J. Immunol., 38, 64-72 (2008)[PubMed]
- Shimada, K., Chen, S., Dempsey, P. W. et al.: The NOD/RIP2 pathway is essential for host defense against Chlamydophila peumoniae lung infection. PLoS Pathog., 5, e1000379 (2009)[PubMed]
- Shaw, P. J., Barr, M. J., Lukens, J. R. et al.: Signaling via the RIP2 adaptor protein in central nervous system-infiltrating dendritic cells promotes inflammation and autoimmunity. Immunity, 34, 75-84 (2011)[PubMed]
著者プロフィール
略歴:2006年 北里大学大学院基礎生命科学研究科博士課程 修了,同年 米国Cedars-Sinai Medical CenterにてPost Doctoral Fellow,2009年 同Research Scientist,2012年 同Assistant Professorを経て,2018年より同Associate Professor.
研究テーマ:感染免疫,アレルギー,動脈硬化,急性肺傷害,川崎病.
© 2018 島田 賢一 Licensed under CC 表示 2.1 日本
