KRAS変異型の非小細胞肺がんに対するTBK1を標的とした新規の治療法の開発
北嶋俊輔・David A. Barbie
(米国Dana-Farber Cancer Institute,Department of Medical Oncology)
email:北嶋俊輔
DOI: 10.7875/first.author.2018.103
Overcoming resistance to dual innate immune and MEK inhibition downstream of KRAS.
Shunsuke Kitajima, Hajime Asahina, Ting Chen, Sujuan Guo, Laura Gutierrez Quiceno, Jillian D. Cavanaugh, Ashley A. Merlino, Shoichiro Tange, Hideki Terai, Jong Wook Kim, Xiaoen Wang, Shan Zhou, Man Xu, Stephen Wang, Zehua Zhu, Tran C. Thai, Chiaki Takahashi, Yujin Wang, Richard Neve, Susanna Stinson, Pablo Tamayo, Hideo Watanabe, Paul T. Kirschmeier, Kwok-Kin Wong, David A. Barbie
Cancer Cell, 34, 439-452.e6 (2018)
KRAS遺伝子変異は非小細胞肺がんにおいて頻繁に観察されるドライバー変異であるが,変異型KRASを標的とする有効な分子標的薬は存在せず,新規の治療法の開発が望まれている.また,KRAS変異型の非小細胞肺がんは,併発するp53遺伝子変異やLKB1遺伝子変異などにより細分化され,それぞれが異なる遺伝子の発現の様式や治療に対する反応性を示すことが知られている.この研究において,筆者らは,KRAS遺伝子変異およびp53遺伝子変異をもつ非小細胞肺がん,および,KRAS遺伝子変異およびLKB1遺伝子変異をもつ非小細胞肺がんについて,マウスモデルおよびヒトの細胞株を用いた比較解析により,KRAS遺伝子変異およびLKB1遺伝子変異をもつ非小細胞肺がんの増殖および生存はRAL経路に依存的なTBK1の活性化に依存すること,さらに,TBK1阻害剤の投与を基盤としたMEK阻害剤およびBET阻害剤との併用により,KRASの下流のMAPK経路あるいはPI3K経路に対する阻害剤が効果を示さないKRAS遺伝子変異およびLKB1遺伝子変異をもつ非小細胞肺がんのモデルにおいて,顕著な腫瘍の抑制および生存期間の延長が認められることを明らかにした.
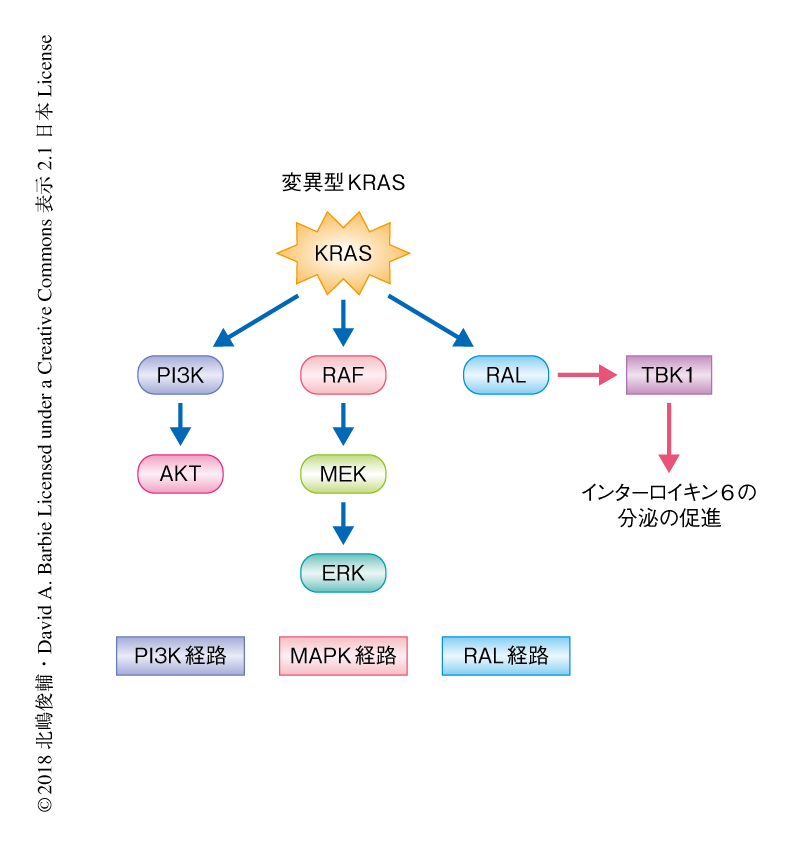
KRAS遺伝子変異は非小細胞肺がんにおいて頻繁に観察され,発がんに直接に寄与するドライバー変異である.しかし,KRASはキナーゼ活性をもたない小分子であるため選択的な阻害剤の開発は難航しており,非小細胞肺がんの代表的なドライバー変異であるEGFR遺伝子変異やALK融合遺伝子と異なり,変異型KRASを標的とした有効な分子標的薬は存在しない1).そのため,KRASの下流の代表的な経路であるMAPK経路あるいはPI3K経路を構成するキナーゼに対する阻害剤を用いた治療法の開発が試みられたが,大きな成功はおさめられていない.一方で,KRASの下流の経路としてRAL経路の活性化も同様に発がんに寄与すると報告されている2).これまでに筆者らは,とくに,RAL経路に依存的なTBK1の活性化がインターロイキン6の分泌の亢進などを介しがん細胞の増殖を促進することを明らかにした3,4)(図1).また,KRAS変異型の非小細胞肺がんに対するMAPK阻害剤およびPI3K阻害剤の併用によりインターロイキン6の分泌がいちじるしく亢進したことから,RAL-TBK1経路がバイパス経路としてがん細胞の生存に寄与することが示唆された.この研究においては,KRAS変異型の非小細胞肺がんに対する効果的な治療法の確立のためには,これまであまり注目されていなかったRAL-TBK1経路の抑制が不可欠であると考え,TBK1阻害剤の投与を組み入れた新規の治療法の開発をめざした.これまでに,TBK1阻害剤の投与によりMAPK経路が活性化することが明らかにされており4),MAPK経路において中心的な役割をはたすMEKに対する阻害剤およびTBK1阻害剤の併用によるKRAS変異型の非小細胞肺がんの治療の効果を,マウスモデルおよびヒトの細胞株を用いて評価した.
近年,KRAS変異型の非小細胞肺がんは併発する遺伝子変異によりいくつかの異なるサブタイプに分類されることが知られている.とくに,がん抑制遺伝子であるp53遺伝子およびLKB1遺伝子の変異が相互排他的に観察され,それぞれは異なる遺伝子の発現の様式を示し,従来の化学療法,分子標的薬,免疫チェックポイント阻害剤に対し異なる感受性を示すと報告されている5,6).これらの背景を考慮し,KRAS遺伝子変異およびp53遺伝子変異をもつKP型の非小細胞肺がん,あるいは,KRAS遺伝子変異およびLKB1遺伝子変異をもつKL型の非小細胞肺がんのモデルを用いて,TBK1阻害剤とMEK阻害剤の併用による治療の効果を評価した.ヒトの細胞株を用いてインターロイキン6の分泌量を比較した結果,KP型の非小細胞肺がんと比較してKL型の非小細胞肺がんにおいて分泌量はいちじるしく多かった.この結果から,KL型の非小細胞肺がんにおいてRAL-TBK1経路の活性の高いことが示唆されたが,実際に,KP型の非小細胞肺がんと比較してKL型の非小細胞肺がんはTBK1阻害剤の処理により細胞の増殖の抑制および細胞死が強くひき起こされた.p53あるいはLKB1の過剰な発現およびCRISPR-Cas9系を用いたゲノム編集による欠損により,インターロイキン6の分泌量およびTBK1阻害剤に対する感受性はおもにLKB1により制御されることが明らかにされた.これらの結果と一致して,TBK1阻害剤とMEK阻害剤の併用はKL型の非小細胞肺がんのマウスモデルに対し顕著な腫瘍の抑制の効果を示した一方,KP型の非小細胞肺がんのマウスモデルに対しては顕著な効果を示さなかった.
がんの分子標的薬に共通する問題として薬剤耐性の獲得があげられる.TBK1阻害剤とMEK阻害剤の併用においても,KL型の非小細胞肺がんのマウスモデルにおいて長期にわたる投与によりがん細胞の再増殖が認められた.そこで,TBK1阻害剤とMEK阻害剤の併用に対する薬剤耐性の機構について解明するため,ヒトの代表的なKL型の非小細胞肺がんの細胞株であるA549細胞を,TBK1阻害剤およびMEK阻害剤の高濃度での存在下において長期にわたり培養することにより,薬剤耐性をもつA549細胞株を作製した.薬剤耐性をもつA549細胞株を薬剤の非存在下にてしばらく培養すると薬剤に対する感受性を完全にとりもどしたことから,薬剤耐性の獲得は,少なくともその初期の過程において,遺伝子変異の獲得ではなく,可逆性をともなうエピジェネティックな制御に依存することが示唆された.そこで,TBK1阻害剤とMEK阻害剤の併用における薬剤耐性に寄与する責任遺伝子を同定するため,RNA-seq法およびヒストンH3のLys27のアセチル化を指標としたChIP-seq法の統合解析により,薬剤の存在下あるいは非存在下において可逆的に発現の変動する659個の遺伝子を同定した.さらに,阻害剤の処理および遺伝子のノックダウンによる解析により,これらのうちとくにIGF1遺伝子およびYAP1遺伝子の発現が上昇しており,IGF1経路およびYAP1経路の活性化がKL型の非小細胞肺がんにおいてTBK1阻害剤とMEK阻害剤の併用における薬剤耐性に寄与することが明らかにされた.
さきに述べたように,TBK1阻害剤とMEK阻害剤の併用に対しKP型の非小細胞肺がん細胞は耐性を示した.そこで,KL型の非小細胞肺がんにおいて薬剤耐性の獲得に寄与する遺伝子が,KP型の非小細胞肺がん細胞において内在的に高く発現している可能性について検討した.その結果,KP型の非小細胞肺がん細胞において,とくにYAP1経路の活性が内在的に高いこと,さらに,遺伝子のノックダウンによる解析により,KP型の非小細胞肺がん細胞においてもYAP1経路の高い活性がTBK1阻害剤とMEK阻害剤の併用に対する薬剤耐性に寄与することが明らかにされた.これらの結果から,YAP1経路の活性化を阻害することにより,TBK1阻害剤とMEK阻害剤の併用に対するKL型の非小細胞肺がんにおける薬剤耐性の獲得,および,KP型の非小細胞肺がんにおける潜在的な薬剤耐性をともに抑制できる可能性が示唆された.
TBK1阻害剤とMEK阻害剤の併用を軸としたより効率的な治療法の確立のため,BET阻害剤の投与がYAP1経路におよぼす影響について解析した.BET阻害剤は,ヒストンH3のLys27のアセチル化を認識し転写を活性化するBRD4などのクロマチンリーダータンパク質を標的とする7).YAP1経路の活性化を抑制する方法としてBET阻害剤を利用した背景としては,KL型の非小細胞肺がんにおける薬剤耐性の獲得においてヒストンH3のLys27のアセチル化の亢進をともなうYAP1経路およびその下流の経路の活性化が観察されること,薬剤耐性をもつ細胞株においてYAP1遺伝子のほかIGF1遺伝子を含むさまざまな遺伝子の発現が上昇しておりBET阻害剤の投与によりそれらの遺伝子の発現を広範に抑制できる可能性のあること,現在のところ治療に使用できる十分な選択性および有効性をもつYAP1阻害剤が存在しないこと,などがあった.実際に,KL型の非小細胞肺がんにおいて,BET阻害剤の投与により,TBK1阻害剤およびMEK阻害剤の投与ののちのYAP1遺伝子およびIGF1遺伝子を含む薬剤耐性に関連する広範な遺伝子の発現の上昇,さらには,薬剤耐性をもつ細胞株の出現が抑制された.また,KP型の非小細胞肺がん細胞においてもBET阻害剤の投与によりYAP1遺伝子の発現が抑制された.そこで,KL型およびKP型の非小細胞肺がんのマウスモデルに対し,TBK1阻害剤,MEK阻害剤,BET阻害剤の併用を試みた.実際のところ,3剤の同時の投与は顕著な体重の減少などの副作用が強かったため,それぞれの薬剤の投与のスケジュールを試行錯誤した結果,BET阻害剤を間歇投与することにより,副作用が軽減されると同時に,KL型の非小細胞肺がんのマウスモデルにおいて薬剤耐性の獲得の抑制および生存期間の延長が認められた.また,さきに述べたように,KP型の非小細胞肺がんのマウスモデルはTBK1阻害剤とMEK阻害剤の併用に対し薬剤耐性を示したが,BET阻害剤の間歇投与の追加により顕著な腫瘍の抑制の効果を示した.
マウスモデルを用いて確立されたTBK1阻害剤,MEK阻害剤,BET阻害剤の併用による治療法の将来的な臨床への応用をめざし,KL型の非小細胞肺がんの患者に由来する腫瘍組織の異種移植モデルを用いて腫瘍の抑制の効果について評価した.ここでは,非常に選択性および有効性の高い新規のTBK1阻害剤およびBET阻害剤を使用した8,9).マウスモデルと同様に,おもに体重の減少を副作用の指標として,長期にわたる投与が可能で治療の効果の高い3剤の併用による治療法の確立をめざした.その結果,TBK1阻害剤とMEK阻害剤の併用と比較して,TBK1阻害剤とMEK阻害剤の併用にくわえBET阻害剤の3日に1回の間歇投与により,有意な腫瘍の抑制および生存期間の延長が認められた.
この研究において,筆者らは,マウスモデルおよびヒトの細胞株を用いた比較解析により,とくにKRAS遺伝子変異およびLKB1遺伝子変異をもつ非小細胞肺がんの増殖および生存は,RAL経路に依存的なTBK1の活性化に依存することを明らかにした.さらに,TBK1阻害剤とMEK阻害剤の併用に対するKRAS遺伝子変異およびLKB1遺伝子変異をもつ非小細胞肺がんにおける薬剤耐性の獲得,および,KRAS遺伝子変異およびp53遺伝子変異をもつ非小細胞肺がんにおける内在的な薬剤耐性の分子機構として,ヒストンH3のLys27のアセチル化の亢進をともなうYAP1遺伝子およびその下流の遺伝子の発現の上昇が重要であること,また,これらの薬剤耐性の機構の抑制にBET阻害剤の間歇投与が有効であることが明らかにされた(図2).KRAS変異型の非小細胞肺がんは生物学的な特性が患者ごとに不均一であり,この研究においては,併発するp53遺伝子変異およびLKB1遺伝子変異に着目しサブタイプごとの生物学的な特性の解明およびそれに対応した治療法の確立をめざしたが,近年,急速に発展するがんゲノミクス解析などにより,今後,さらなる細分化が進むと考えられる.現在まで,KRAS変異型の非小細胞肺がんに対する特異的な治療法は確立されていないが,生物学的な特異性を基盤とした正確なサブタイプの分類およびそれに対応した治療法の開発が期待される.また近年,既存の化学療法,分子標的薬,免疫チェックポイント阻害剤などをさまざまに組み合わせた治療法がさかんに開発されているが,この研究においては,同様の組合せであっても投薬のスケジュールにより治療の成績は大きく異なることが示された.BET阻害剤は副作用の強さから臨床において期待されたほどの成果が得られていないが,定期的な間歇投与により,種々のがん治療薬に対する薬剤耐性の獲得の初期の過程に寄与すると考えられるエピジェネティックな制御を抑制することで治療の効果が増強されることが期待される.

略歴:2010年 京都大学大学院医学研究科博士課程 修了,同年 金沢大学がん進展制御研究所 博士研究員,2013年 同 特任助教を経て,2015年より米国Dana-Farber Cancer Institute博士研究員.
研究テーマ:がん遺伝子およびがん抑制遺伝子による自然免疫系のシグナルの制御.
関心事:腫瘍組織における遺伝子の異常と微小環境の組成との関連.
David A. Barbie
米国Harvard Medical School助教.
© 2018 北嶋俊輔・David A. Barbie Licensed under CC 表示 2.1 日本
(米国Dana-Farber Cancer Institute,Department of Medical Oncology)
email:北嶋俊輔
DOI: 10.7875/first.author.2018.103
Overcoming resistance to dual innate immune and MEK inhibition downstream of KRAS.
Shunsuke Kitajima, Hajime Asahina, Ting Chen, Sujuan Guo, Laura Gutierrez Quiceno, Jillian D. Cavanaugh, Ashley A. Merlino, Shoichiro Tange, Hideki Terai, Jong Wook Kim, Xiaoen Wang, Shan Zhou, Man Xu, Stephen Wang, Zehua Zhu, Tran C. Thai, Chiaki Takahashi, Yujin Wang, Richard Neve, Susanna Stinson, Pablo Tamayo, Hideo Watanabe, Paul T. Kirschmeier, Kwok-Kin Wong, David A. Barbie
Cancer Cell, 34, 439-452.e6 (2018)
この論文に出現する遺伝子・タンパク質のUniprot ID
KRAS(P01116), TBK1(Q9UHD2), MEK, p53(P04637), LKB1(Q15831), RAL(P11233), MAPK, PI3K, EGFR(P00533), ALK(Q9UM73), インターロイキン6(P05231), IGF1(P05019), YAP1(P46937), ヒストンH3, BRD4(O60885), YAP(P46937)
要 約
KRAS遺伝子変異は非小細胞肺がんにおいて頻繁に観察されるドライバー変異であるが,変異型KRASを標的とする有効な分子標的薬は存在せず,新規の治療法の開発が望まれている.また,KRAS変異型の非小細胞肺がんは,併発するp53遺伝子変異やLKB1遺伝子変異などにより細分化され,それぞれが異なる遺伝子の発現の様式や治療に対する反応性を示すことが知られている.この研究において,筆者らは,KRAS遺伝子変異およびp53遺伝子変異をもつ非小細胞肺がん,および,KRAS遺伝子変異およびLKB1遺伝子変異をもつ非小細胞肺がんについて,マウスモデルおよびヒトの細胞株を用いた比較解析により,KRAS遺伝子変異およびLKB1遺伝子変異をもつ非小細胞肺がんの増殖および生存はRAL経路に依存的なTBK1の活性化に依存すること,さらに,TBK1阻害剤の投与を基盤としたMEK阻害剤およびBET阻害剤との併用により,KRASの下流のMAPK経路あるいはPI3K経路に対する阻害剤が効果を示さないKRAS遺伝子変異およびLKB1遺伝子変異をもつ非小細胞肺がんのモデルにおいて,顕著な腫瘍の抑制および生存期間の延長が認められることを明らかにした.
はじめに
KRAS遺伝子変異は非小細胞肺がんにおいて頻繁に観察され,発がんに直接に寄与するドライバー変異である.しかし,KRASはキナーゼ活性をもたない小分子であるため選択的な阻害剤の開発は難航しており,非小細胞肺がんの代表的なドライバー変異であるEGFR遺伝子変異やALK融合遺伝子と異なり,変異型KRASを標的とした有効な分子標的薬は存在しない1).そのため,KRASの下流の代表的な経路であるMAPK経路あるいはPI3K経路を構成するキナーゼに対する阻害剤を用いた治療法の開発が試みられたが,大きな成功はおさめられていない.一方で,KRASの下流の経路としてRAL経路の活性化も同様に発がんに寄与すると報告されている2).これまでに筆者らは,とくに,RAL経路に依存的なTBK1の活性化がインターロイキン6の分泌の亢進などを介しがん細胞の増殖を促進することを明らかにした3,4)(図1).また,KRAS変異型の非小細胞肺がんに対するMAPK阻害剤およびPI3K阻害剤の併用によりインターロイキン6の分泌がいちじるしく亢進したことから,RAL-TBK1経路がバイパス経路としてがん細胞の生存に寄与することが示唆された.この研究においては,KRAS変異型の非小細胞肺がんに対する効果的な治療法の確立のためには,これまであまり注目されていなかったRAL-TBK1経路の抑制が不可欠であると考え,TBK1阻害剤の投与を組み入れた新規の治療法の開発をめざした.これまでに,TBK1阻害剤の投与によりMAPK経路が活性化することが明らかにされており4),MAPK経路において中心的な役割をはたすMEKに対する阻害剤およびTBK1阻害剤の併用によるKRAS変異型の非小細胞肺がんの治療の効果を,マウスモデルおよびヒトの細胞株を用いて評価した.
1.TBK1阻害剤とMEK阻害剤の併用はKRAS遺伝子変異およびLKB1遺伝子変異をもつ非小細胞肺がんに対し顕著な効果を示す
近年,KRAS変異型の非小細胞肺がんは併発する遺伝子変異によりいくつかの異なるサブタイプに分類されることが知られている.とくに,がん抑制遺伝子であるp53遺伝子およびLKB1遺伝子の変異が相互排他的に観察され,それぞれは異なる遺伝子の発現の様式を示し,従来の化学療法,分子標的薬,免疫チェックポイント阻害剤に対し異なる感受性を示すと報告されている5,6).これらの背景を考慮し,KRAS遺伝子変異およびp53遺伝子変異をもつKP型の非小細胞肺がん,あるいは,KRAS遺伝子変異およびLKB1遺伝子変異をもつKL型の非小細胞肺がんのモデルを用いて,TBK1阻害剤とMEK阻害剤の併用による治療の効果を評価した.ヒトの細胞株を用いてインターロイキン6の分泌量を比較した結果,KP型の非小細胞肺がんと比較してKL型の非小細胞肺がんにおいて分泌量はいちじるしく多かった.この結果から,KL型の非小細胞肺がんにおいてRAL-TBK1経路の活性の高いことが示唆されたが,実際に,KP型の非小細胞肺がんと比較してKL型の非小細胞肺がんはTBK1阻害剤の処理により細胞の増殖の抑制および細胞死が強くひき起こされた.p53あるいはLKB1の過剰な発現およびCRISPR-Cas9系を用いたゲノム編集による欠損により,インターロイキン6の分泌量およびTBK1阻害剤に対する感受性はおもにLKB1により制御されることが明らかにされた.これらの結果と一致して,TBK1阻害剤とMEK阻害剤の併用はKL型の非小細胞肺がんのマウスモデルに対し顕著な腫瘍の抑制の効果を示した一方,KP型の非小細胞肺がんのマウスモデルに対しては顕著な効果を示さなかった.
2.KL型の非小細胞肺がんにおける薬剤耐性の獲得においてIGF1経路およびYAP1経路の活性化が寄与する
がんの分子標的薬に共通する問題として薬剤耐性の獲得があげられる.TBK1阻害剤とMEK阻害剤の併用においても,KL型の非小細胞肺がんのマウスモデルにおいて長期にわたる投与によりがん細胞の再増殖が認められた.そこで,TBK1阻害剤とMEK阻害剤の併用に対する薬剤耐性の機構について解明するため,ヒトの代表的なKL型の非小細胞肺がんの細胞株であるA549細胞を,TBK1阻害剤およびMEK阻害剤の高濃度での存在下において長期にわたり培養することにより,薬剤耐性をもつA549細胞株を作製した.薬剤耐性をもつA549細胞株を薬剤の非存在下にてしばらく培養すると薬剤に対する感受性を完全にとりもどしたことから,薬剤耐性の獲得は,少なくともその初期の過程において,遺伝子変異の獲得ではなく,可逆性をともなうエピジェネティックな制御に依存することが示唆された.そこで,TBK1阻害剤とMEK阻害剤の併用における薬剤耐性に寄与する責任遺伝子を同定するため,RNA-seq法およびヒストンH3のLys27のアセチル化を指標としたChIP-seq法の統合解析により,薬剤の存在下あるいは非存在下において可逆的に発現の変動する659個の遺伝子を同定した.さらに,阻害剤の処理および遺伝子のノックダウンによる解析により,これらのうちとくにIGF1遺伝子およびYAP1遺伝子の発現が上昇しており,IGF1経路およびYAP1経路の活性化がKL型の非小細胞肺がんにおいてTBK1阻害剤とMEK阻害剤の併用における薬剤耐性に寄与することが明らかにされた.
3.KP型の非小細胞肺がんにおいてYAP1経路の活性が高い
さきに述べたように,TBK1阻害剤とMEK阻害剤の併用に対しKP型の非小細胞肺がん細胞は耐性を示した.そこで,KL型の非小細胞肺がんにおいて薬剤耐性の獲得に寄与する遺伝子が,KP型の非小細胞肺がん細胞において内在的に高く発現している可能性について検討した.その結果,KP型の非小細胞肺がん細胞において,とくにYAP1経路の活性が内在的に高いこと,さらに,遺伝子のノックダウンによる解析により,KP型の非小細胞肺がん細胞においてもYAP1経路の高い活性がTBK1阻害剤とMEK阻害剤の併用に対する薬剤耐性に寄与することが明らかにされた.これらの結果から,YAP1経路の活性化を阻害することにより,TBK1阻害剤とMEK阻害剤の併用に対するKL型の非小細胞肺がんにおける薬剤耐性の獲得,および,KP型の非小細胞肺がんにおける潜在的な薬剤耐性をともに抑制できる可能性が示唆された.
4.KL型およびKP型の非小細胞肺がんのマウスモデルにおいてTBK1阻害剤,MEK阻害剤,BET阻害剤の併用が有効である
TBK1阻害剤とMEK阻害剤の併用を軸としたより効率的な治療法の確立のため,BET阻害剤の投与がYAP1経路におよぼす影響について解析した.BET阻害剤は,ヒストンH3のLys27のアセチル化を認識し転写を活性化するBRD4などのクロマチンリーダータンパク質を標的とする7).YAP1経路の活性化を抑制する方法としてBET阻害剤を利用した背景としては,KL型の非小細胞肺がんにおける薬剤耐性の獲得においてヒストンH3のLys27のアセチル化の亢進をともなうYAP1経路およびその下流の経路の活性化が観察されること,薬剤耐性をもつ細胞株においてYAP1遺伝子のほかIGF1遺伝子を含むさまざまな遺伝子の発現が上昇しておりBET阻害剤の投与によりそれらの遺伝子の発現を広範に抑制できる可能性のあること,現在のところ治療に使用できる十分な選択性および有効性をもつYAP1阻害剤が存在しないこと,などがあった.実際に,KL型の非小細胞肺がんにおいて,BET阻害剤の投与により,TBK1阻害剤およびMEK阻害剤の投与ののちのYAP1遺伝子およびIGF1遺伝子を含む薬剤耐性に関連する広範な遺伝子の発現の上昇,さらには,薬剤耐性をもつ細胞株の出現が抑制された.また,KP型の非小細胞肺がん細胞においてもBET阻害剤の投与によりYAP1遺伝子の発現が抑制された.そこで,KL型およびKP型の非小細胞肺がんのマウスモデルに対し,TBK1阻害剤,MEK阻害剤,BET阻害剤の併用を試みた.実際のところ,3剤の同時の投与は顕著な体重の減少などの副作用が強かったため,それぞれの薬剤の投与のスケジュールを試行錯誤した結果,BET阻害剤を間歇投与することにより,副作用が軽減されると同時に,KL型の非小細胞肺がんのマウスモデルにおいて薬剤耐性の獲得の抑制および生存期間の延長が認められた.また,さきに述べたように,KP型の非小細胞肺がんのマウスモデルはTBK1阻害剤とMEK阻害剤の併用に対し薬剤耐性を示したが,BET阻害剤の間歇投与の追加により顕著な腫瘍の抑制の効果を示した.
5.KL型の非小細胞肺がんの患者に由来するモデルにおいてTBK1阻害剤,MEK阻害剤,BET阻害剤の併用が有効である
マウスモデルを用いて確立されたTBK1阻害剤,MEK阻害剤,BET阻害剤の併用による治療法の将来的な臨床への応用をめざし,KL型の非小細胞肺がんの患者に由来する腫瘍組織の異種移植モデルを用いて腫瘍の抑制の効果について評価した.ここでは,非常に選択性および有効性の高い新規のTBK1阻害剤およびBET阻害剤を使用した8,9).マウスモデルと同様に,おもに体重の減少を副作用の指標として,長期にわたる投与が可能で治療の効果の高い3剤の併用による治療法の確立をめざした.その結果,TBK1阻害剤とMEK阻害剤の併用と比較して,TBK1阻害剤とMEK阻害剤の併用にくわえBET阻害剤の3日に1回の間歇投与により,有意な腫瘍の抑制および生存期間の延長が認められた.
おわりに
この研究において,筆者らは,マウスモデルおよびヒトの細胞株を用いた比較解析により,とくにKRAS遺伝子変異およびLKB1遺伝子変異をもつ非小細胞肺がんの増殖および生存は,RAL経路に依存的なTBK1の活性化に依存することを明らかにした.さらに,TBK1阻害剤とMEK阻害剤の併用に対するKRAS遺伝子変異およびLKB1遺伝子変異をもつ非小細胞肺がんにおける薬剤耐性の獲得,および,KRAS遺伝子変異およびp53遺伝子変異をもつ非小細胞肺がんにおける内在的な薬剤耐性の分子機構として,ヒストンH3のLys27のアセチル化の亢進をともなうYAP1遺伝子およびその下流の遺伝子の発現の上昇が重要であること,また,これらの薬剤耐性の機構の抑制にBET阻害剤の間歇投与が有効であることが明らかにされた(図2).KRAS変異型の非小細胞肺がんは生物学的な特性が患者ごとに不均一であり,この研究においては,併発するp53遺伝子変異およびLKB1遺伝子変異に着目しサブタイプごとの生物学的な特性の解明およびそれに対応した治療法の確立をめざしたが,近年,急速に発展するがんゲノミクス解析などにより,今後,さらなる細分化が進むと考えられる.現在まで,KRAS変異型の非小細胞肺がんに対する特異的な治療法は確立されていないが,生物学的な特異性を基盤とした正確なサブタイプの分類およびそれに対応した治療法の開発が期待される.また近年,既存の化学療法,分子標的薬,免疫チェックポイント阻害剤などをさまざまに組み合わせた治療法がさかんに開発されているが,この研究においては,同様の組合せであっても投薬のスケジュールにより治療の成績は大きく異なることが示された.BET阻害剤は副作用の強さから臨床において期待されたほどの成果が得られていないが,定期的な間歇投与により,種々のがん治療薬に対する薬剤耐性の獲得の初期の過程に寄与すると考えられるエピジェネティックな制御を抑制することで治療の効果が増強されることが期待される.
文 献
- Stephen, A. G., Esposito, D., Bagni, R. K. et al.: Dragging Ras back in the ring. Cancer Cell, 25, 272-281 (2014)[PubMed]
- Bodemann, B. O. & White, M. A.: Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nat. Rev. Cancer, 8, 133-140 (2008)[PubMed]
- Barbie, D. A., Tamayo, P., Boehm, J. S. et al.: Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature, 462, 108-112 (2009)[PubMed]
- Zhu, Z., Aref, A. R., Cohoon, T. J. et al.: Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov., 4, 452-465 (2014)[PubMed]
- Skoulidis, F., Byers, L. A., Diao, L. et al.: Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov., 5, 860-877 (2015)[PubMed]
- Skoulidis, F., Goldberg, M. E., Greenawalt, D. M. et al.: STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov., 8, 822-835 (2018)[PubMed]
- Shi, J. & Vakoc, C. R.: The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell, 54, 728-736 (2014)[PubMed]
- Jenkins, R. W., Aref, A. R., Lizotte, P. H. et al.: Ex vivo profiling of PD-1 blockade using organotypic tumor spheroids. Cancer Discov., 8, 196-215 (2018)[PubMed]
- Shi, X., Mihaylova, V. T., Kuruvilla, L. et al.: Loss of TRIM33 causes resistance to BET bromodomain inhibitors through MYC- and TGF-β-dependent mechanisms. Proc. Natl. Acad. Sci. USA, 113, E4558-E4566 (2016)[PubMed]
活用したデータベースにかかわるキーワードと統合TVへのリンク
著者プロフィール
略歴:2010年 京都大学大学院医学研究科博士課程 修了,同年 金沢大学がん進展制御研究所 博士研究員,2013年 同 特任助教を経て,2015年より米国Dana-Farber Cancer Institute博士研究員.
研究テーマ:がん遺伝子およびがん抑制遺伝子による自然免疫系のシグナルの制御.
関心事:腫瘍組織における遺伝子の異常と微小環境の組成との関連.
David A. Barbie
米国Harvard Medical School助教.
© 2018 北嶋俊輔・David A. Barbie Licensed under CC 表示 2.1 日本
