高CO2はアポトーシスの制御機構とは異なるカスパーゼ7の活性化を介して気道平滑筋を収縮させる
重村雅彦・Jacob I. Sznajder
(米国Northwestern大学Feinberg School of Medicine,Division of Pulmonary and Critical Care)
email:重村雅彦
DOI: 10.7875/first.author.2018.096
Hypercapnia increases airway smooth muscle contractility via caspase-7-mediated miR-133a-RhoA signaling.
Masahiko Shigemura , Emilia Lecuona, Martín Angulo, Tetsuya Homma, Diego A. Rodríguez, Francisco J. Gonzalez-Gonzalez, Lynn C. Welch, Luciano Amarelle, Seok-Jo Kim, Naftali Kaminski, G. R. Scott Budinger, Julian Solway, Jacob I. Sznajder
Science Translational Medicine, 10, eaat1662 (2018)
呼吸不全をともなう慢性閉塞性肺疾患の患者は高CO2血症をともなうことが知られている.高CO2血症は慢性閉塞性肺疾患の重症度の指標のひとつとされているが,高CO2が慢性閉塞性肺疾患の病態におよぼす役割についてはほとんど知られていない.この研究において,筆者らは,CO2がシグナル伝達分子としてマウスおよびヒトの気道平滑筋細胞に作用することを明らかにした.高CO2条件においては,気道平滑筋細胞がアポトーシスの制御機構とは異なるカスパーゼ7の活性化を介して収縮し,その結果,マウスにおいて気道の抵抗を上昇させた.一方,慢性閉塞性肺疾患のコホート研究を用いて,高CO2血症をともなう患者において気道の抵抗および呼吸の抵抗が上昇していること,また,非侵襲的な陽圧換気療法により高CO2血症が改善された症例において呼吸の抵抗が低下することが見い出された.この研究により,CO2の気道の病態生理への関与が明らかにされた.高CO2血症を呈する慢性閉塞性肺疾患の患者においては,高CO2により気道が収縮し,その結果,高CO2血症が進行して呼吸不全が悪化する可能性が示唆された.
O2,NO,CO2などのガス状の分子はシグナル伝達分子としてさまざまな生理機能をもつことが知られている.CO2は生体においてすみやかにH+とHCO3-に変換されることから,これまで,CO2によるシグナル伝達はおもにH+のはたらきによりひき起こされると考えられてきた.これに対し,最近,肺や骨格筋などの組織において,CO2がシグナル伝達分子として直接的にはたらくことが示唆されている1).
高CO2血症は肺胞の低換気をともなう慢性閉塞性肺疾患など,種々の呼吸器疾患に認められる.慢性閉塞性肺疾患により世界で年間300万人が死亡しており,2030年までには死因の第3位になると予想されている.慢性的な高CO2血症は慢性閉塞性肺疾患の死亡率の上昇と関連する2).最近,高CO2血症を呈する慢性閉塞性肺疾患の患者において,非侵襲的な陽圧換気療法による介入が死亡率の低下やQOL(quality of life)の改善と関連すると報告された3,4).
この研究においては,筆者らは,高CO2が気道の病態生理になんらかの影響をおよぼすという仮説をたて,高CO2の影響および機序をin vitroのモデルおよびマウスのin vivoのモデル,さらには,慢性閉塞性肺疾患のコホート研究を用いて検討した.
高CO2が肺におよぼす影響について網羅的に解析するため,高CO2かつ正常なO2に曝露されたマウスの肺組織においてmRNAおよびmiRNAに対しマイクロアレイ解析を実施した.その結果,3日間の曝露では1017個のmRNA,7日間の曝露では2959個のmRNAの発現に変化が認められた.また,マイクロアレイのデータのパスウェイ解析により,気道平滑筋の収縮に関連するシグナル伝達経路がもっとも活性化されていることが示唆された.そこで,気道平滑筋の収縮に関連するシグナル伝達経路を推定するため,mRNAおよびmiRNAのマイクロアレイのデータを用いて遺伝子ネットワーク解析を実施したところ,miR-133aを介したRhoA-ROCK-ミオシン軽鎖ホスファターゼ-ミオシン軽鎖というシグナル伝達経路が示唆された.
高CO2に曝露されたマウスの肺組織のマイクロアレイ解析から,高CO2は気道平滑筋を収縮させることが示唆された.この仮説について検討するため,マウスおよびヒトの気道平滑筋細胞を用いて,高CO2かつ正常なO2に曝露された平滑筋細胞において,アセチルコリンによりひき起こされる収縮とCO2の濃度およびpHとの関係について検討した.その結果,気道平滑筋細胞はCO2の濃度および曝露の期間に依存して収縮したが,pHの酸性化とは関連はなかった.
高CO2により気道平滑筋細胞が収縮する機序について検討した.マウスの肺組織のマイクロアレイ解析において,miR-133aを介したRhoA-ROCK-ミオシン軽鎖ホスファターゼ-ミオシン軽鎖というシグナル伝達経路が示唆された.これまでに,気道平滑筋細胞の収縮はミオシン軽鎖のリン酸化により制御され,また,ミオシン軽鎖のリン酸化はRhoA-ROCK-ミオシン軽鎖ホスファターゼというシグナル伝達経路により制御されることが報告されていた5).さらに最近,miR-133aはRhoAの発現および活性化を制御することも明らかにされた6).そこで,高CO2条件において気道平滑筋細胞におけるミオシン軽鎖のリン酸化,および,RhoAおよびmiR-133aの発現について検討したところ,高CO2がmiR-133aの発現を抑制し,その結果,RhoAの発現が上昇してミオシン軽鎖のリン酸化が促進されることがマウスおよびヒトの細胞において確認された.
高CO2によりmiR-133aの発現が抑制される機序について検討するため,転写因子結合データベースにおいてmiR-133a遺伝子のプロモーター領域に結合する転写因子を探索したところ,YY1およびMEF2Dが見い出された.マウスの気道平滑筋細胞においてYY1あるいはMEF2DをノックダウンしてmiR-133aの発現について検討したところ,MEF2DがmiR-133aの発現を制御する転写因子であることが明らかにされた.そこで,染色およびウェスタンブロット法により高CO2条件におけるMEF2Dの発現について検討したところ,MEF2Dは気道平滑筋細胞の核の周辺に局在し,核におけるタンパク質量は減少していた.クロマチン免疫沈降法によりmiR-133a遺伝子のプロモーター領域へのMEF2Dの結合について検討したところ,高CO2条件においてMEF2Dの結合は減少していた.以上のことから,高CO2によりMEF2Dの核における局在が低下し,その結果,miR-133a遺伝子のプロモーター領域への結合が減少することによりmiR-133aの発現は抑制されると考えられた.
MEF2Dのウェスタンブロット法による解析において,タンパク質量の減少ともにタンパク質の断片化も観察された.これまでに,MEF2Dの断片化においてカスパーゼの関与が報告されていた.高CO2条件におけるカスパーゼによるMEF2Dの断片化について検討するため,カスパーゼの阻害剤を用いてMEF2Dの断片化およびmiR-133aの発現について検討したところ,高CO2によるMEF2Dの断片化およびmiR-133の発現の抑制は改善された.高CO2により活性化するカスパーゼを調べたところ,マウスの気道平滑筋細胞においてカスパーゼ7のみが高CO2により活性化されていた.カスパーゼ7をノックダウンしてMEF2Dの断片化およびmiR-133aの発現について検討したところ,高CO2によるMEF2Dの断片化およびmiR-133の発現の抑制は改善された.カスパーゼは細胞においてアポトーシスを制御することで知られている7).しかし,高CO2条件において気道平滑筋細胞のアポトーシスは認められなかった.以上のことから,高CO2はアポトーシスの制御機構とは異なるカスパーゼ7の活性化を介して気道平滑筋細胞を収縮させると考えられた.
カスパーゼの活性はCa2+依存性のシステインプロテアーゼであるカルパインにより制御されることが報告されていた8).マウスの気道平滑筋細胞におけるカルパインの活性について検討したところ,高CO2により一時的な上昇が観察された.高CO2条件において,組織に普遍的に発現するカルパイン1およびカルパイン2をノックダウンして気道平滑筋細胞におけるカスパーゼ7の活性について検討したところ,カルパイン1のノックダウンによりカスパーゼ7の活性は抑制された.一方,高CO2はマウスの気道平滑筋細胞において細胞におけるCa2+の濃度を上昇させ,また,Ca2+のキレート剤は高CO2によるカルパインの活性の上昇を抑制した.以上のことから,高CO2は気道平滑筋細胞におけるCa2+の濃度を上昇させ,カルパインの活性を介してカスパーゼ7を活性化させると考えられた(図1).
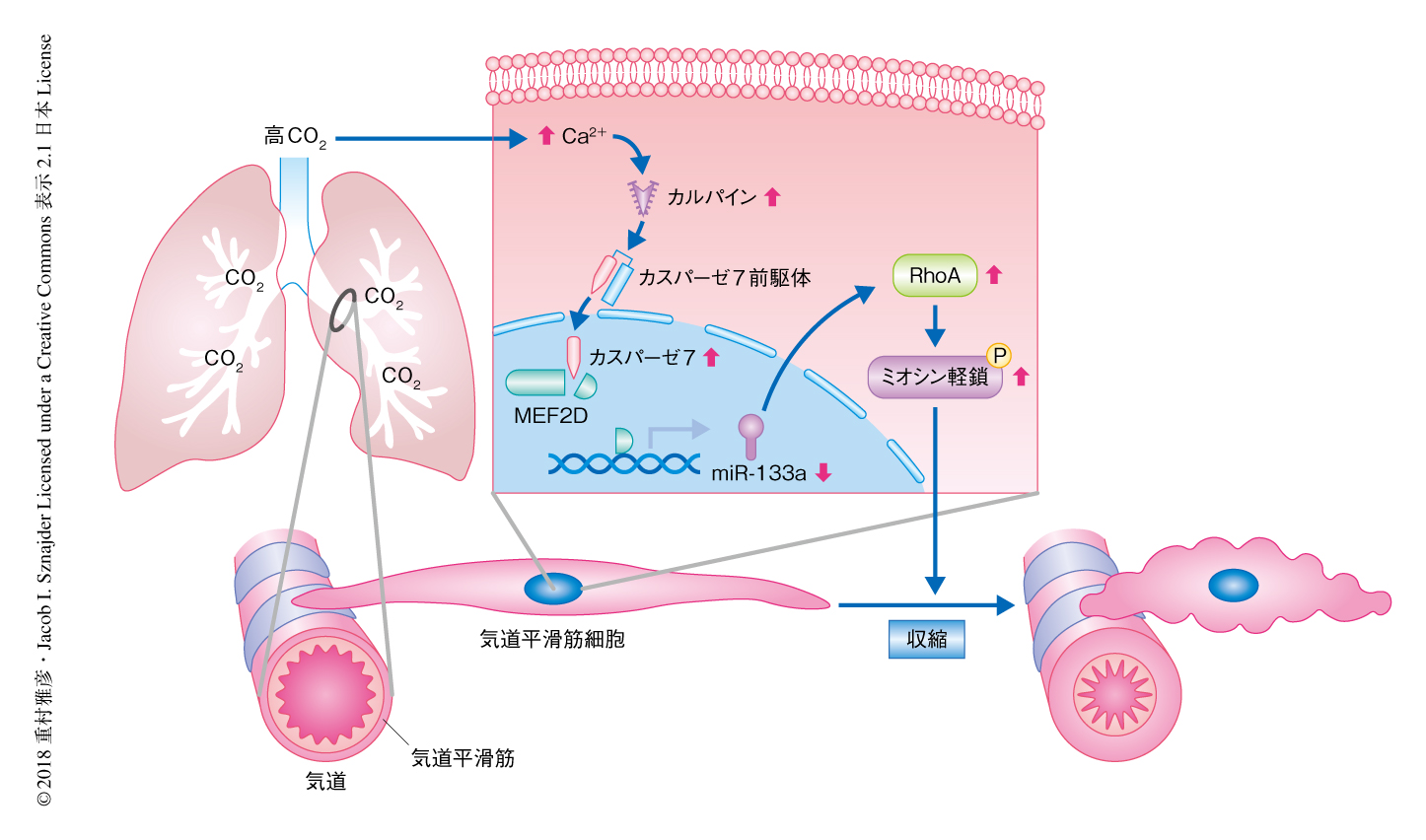
in vitroの実験系において認められた高CO2による気道平滑筋細胞の収縮の機序が,マウスのin vivoのモデルにおいても確認されるか検討した.高CO2かつ正常なO2に曝露された野生型のマウスの気道の収縮および気道の抵抗について検討したところ,気道の収縮はCO2の曝露の期間と比例して上昇し,また,気道の抵抗も上昇した.また,高CO2に曝露されたマウスの肺組織においては,miR-133aの発現は低下,RhoAの発現は上昇,ミオシン軽鎖はリン酸化されていた.一方,カスパーゼ7のノックアウトマウスにおいては,高CO2による気道の収縮および気道の抵抗の上昇は抑制され,また,高CO2によりひき起こされるmiR-133a-RhoA-ROCK-ミオシン軽鎖ホスファターゼ-ミオシン軽鎖というシグナル伝達経路の下流のシグナルも抑制されていた.これに対し,経気道的にmiR-133aの阻害剤を導入したカスパーゼ7のノックアウトマウスにおいては,野生型のマウスと同様に,高CO2により気道が収縮し,高CO2によりひき起こされる下流のシグナルも認められた.
高CO2がヒトにおいても気道の収縮に影響をおよぼすのかどうか検討するため,慢性閉塞性肺疾患のコホート研究を用いて,高CO2血症を呈する重症の患者および高CO2血症を呈さない重症の患者を対象に,気道および呼吸の抵抗を測定した.その結果,高CO2血症をともなう患者においては気道および呼吸の抵抗が上昇し,また,非侵襲的な陽圧換気療法による介入により高CO2血症が改善された症例においては呼吸の抵抗が低下した.
この研究において,筆者らは,CO2がシグナル伝達分子として気道平滑筋細胞の収縮に影響をおよぼすことを明らかにした.高CO2は気道平滑筋に有害であることが示唆されたが,高CO2血症を呈する慢性閉塞性肺疾患の患者において,高CO2を容認するか否かについては議論がある.その背景のひとつに,急性肺障害/急性呼吸窮迫症候群の患者においては,呼吸の管理において高CO2血症を容認するという概念が広く普及していることがある9).これは,肺の過膨張による正常な肺の障害を保護する目的で推奨された呼吸の管理法であるが,一方で,高CO2血症にともなう呼吸性のアシドーシスが抗炎症応答にも寄与することが示唆されており,このことから,治療のための高CO2血症という治療戦略も提唱されている.近年になり,慢性閉塞性肺疾患の患者において高CO2血症への介入が検討され3,4),その有効性が議論されている.この研究の成果は,これらの臨床研究を分子生物学的な観点からサポートする知見であり意義深い.
略歴:2011年 北海道大学大学院医学研究科博士課程 修了,2013年より米国Northwestern大学Feinberg School of Medicine博士研究員.
研究テーマ:CO2による生態制御の生物学.
関心事:生物の代謝機構におけるCO2の役割およびセンサー分子の探索.
Jacob I. Sznajder
米国Northwestern大学Feinberg School of Medicine教授.
研究室URL:https://labs.feinberg.northwestern.edu/sznajder/
© 2018 重村雅彦・Jacob I. Sznajder Licensed under CC 表示 2.1 日本
(米国Northwestern大学Feinberg School of Medicine,Division of Pulmonary and Critical Care)
email:重村雅彦
DOI: 10.7875/first.author.2018.096
Hypercapnia increases airway smooth muscle contractility via caspase-7-mediated miR-133a-RhoA signaling.
Masahiko Shigemura , Emilia Lecuona, Martín Angulo, Tetsuya Homma, Diego A. Rodríguez, Francisco J. Gonzalez-Gonzalez, Lynn C. Welch, Luciano Amarelle, Seok-Jo Kim, Naftali Kaminski, G. R. Scott Budinger, Julian Solway, Jacob I. Sznajder
Science Translational Medicine, 10, eaat1662 (2018)
要 約
呼吸不全をともなう慢性閉塞性肺疾患の患者は高CO2血症をともなうことが知られている.高CO2血症は慢性閉塞性肺疾患の重症度の指標のひとつとされているが,高CO2が慢性閉塞性肺疾患の病態におよぼす役割についてはほとんど知られていない.この研究において,筆者らは,CO2がシグナル伝達分子としてマウスおよびヒトの気道平滑筋細胞に作用することを明らかにした.高CO2条件においては,気道平滑筋細胞がアポトーシスの制御機構とは異なるカスパーゼ7の活性化を介して収縮し,その結果,マウスにおいて気道の抵抗を上昇させた.一方,慢性閉塞性肺疾患のコホート研究を用いて,高CO2血症をともなう患者において気道の抵抗および呼吸の抵抗が上昇していること,また,非侵襲的な陽圧換気療法により高CO2血症が改善された症例において呼吸の抵抗が低下することが見い出された.この研究により,CO2の気道の病態生理への関与が明らかにされた.高CO2血症を呈する慢性閉塞性肺疾患の患者においては,高CO2により気道が収縮し,その結果,高CO2血症が進行して呼吸不全が悪化する可能性が示唆された.
はじめに
O2,NO,CO2などのガス状の分子はシグナル伝達分子としてさまざまな生理機能をもつことが知られている.CO2は生体においてすみやかにH+とHCO3-に変換されることから,これまで,CO2によるシグナル伝達はおもにH+のはたらきによりひき起こされると考えられてきた.これに対し,最近,肺や骨格筋などの組織において,CO2がシグナル伝達分子として直接的にはたらくことが示唆されている1).
高CO2血症は肺胞の低換気をともなう慢性閉塞性肺疾患など,種々の呼吸器疾患に認められる.慢性閉塞性肺疾患により世界で年間300万人が死亡しており,2030年までには死因の第3位になると予想されている.慢性的な高CO2血症は慢性閉塞性肺疾患の死亡率の上昇と関連する2).最近,高CO2血症を呈する慢性閉塞性肺疾患の患者において,非侵襲的な陽圧換気療法による介入が死亡率の低下やQOL(quality of life)の改善と関連すると報告された3,4).
この研究においては,筆者らは,高CO2が気道の病態生理になんらかの影響をおよぼすという仮説をたて,高CO2の影響および機序をin vitroのモデルおよびマウスのin vivoのモデル,さらには,慢性閉塞性肺疾患のコホート研究を用いて検討した.
1.高CO2に曝露されたマウスの肺組織において気道平滑筋の収縮に関連する遺伝子の発現が変化する
高CO2が肺におよぼす影響について網羅的に解析するため,高CO2かつ正常なO2に曝露されたマウスの肺組織においてmRNAおよびmiRNAに対しマイクロアレイ解析を実施した.その結果,3日間の曝露では1017個のmRNA,7日間の曝露では2959個のmRNAの発現に変化が認められた.また,マイクロアレイのデータのパスウェイ解析により,気道平滑筋の収縮に関連するシグナル伝達経路がもっとも活性化されていることが示唆された.そこで,気道平滑筋の収縮に関連するシグナル伝達経路を推定するため,mRNAおよびmiRNAのマイクロアレイのデータを用いて遺伝子ネットワーク解析を実施したところ,miR-133aを介したRhoA-ROCK-ミオシン軽鎖ホスファターゼ-ミオシン軽鎖というシグナル伝達経路が示唆された.
2.高CO2はアポトーシスの制御機構とは異なるカスパーゼ7の活性化を介して気道平滑筋細胞を収縮させる
高CO2に曝露されたマウスの肺組織のマイクロアレイ解析から,高CO2は気道平滑筋を収縮させることが示唆された.この仮説について検討するため,マウスおよびヒトの気道平滑筋細胞を用いて,高CO2かつ正常なO2に曝露された平滑筋細胞において,アセチルコリンによりひき起こされる収縮とCO2の濃度およびpHとの関係について検討した.その結果,気道平滑筋細胞はCO2の濃度および曝露の期間に依存して収縮したが,pHの酸性化とは関連はなかった.
高CO2により気道平滑筋細胞が収縮する機序について検討した.マウスの肺組織のマイクロアレイ解析において,miR-133aを介したRhoA-ROCK-ミオシン軽鎖ホスファターゼ-ミオシン軽鎖というシグナル伝達経路が示唆された.これまでに,気道平滑筋細胞の収縮はミオシン軽鎖のリン酸化により制御され,また,ミオシン軽鎖のリン酸化はRhoA-ROCK-ミオシン軽鎖ホスファターゼというシグナル伝達経路により制御されることが報告されていた5).さらに最近,miR-133aはRhoAの発現および活性化を制御することも明らかにされた6).そこで,高CO2条件において気道平滑筋細胞におけるミオシン軽鎖のリン酸化,および,RhoAおよびmiR-133aの発現について検討したところ,高CO2がmiR-133aの発現を抑制し,その結果,RhoAの発現が上昇してミオシン軽鎖のリン酸化が促進されることがマウスおよびヒトの細胞において確認された.
高CO2によりmiR-133aの発現が抑制される機序について検討するため,転写因子結合データベースにおいてmiR-133a遺伝子のプロモーター領域に結合する転写因子を探索したところ,YY1およびMEF2Dが見い出された.マウスの気道平滑筋細胞においてYY1あるいはMEF2DをノックダウンしてmiR-133aの発現について検討したところ,MEF2DがmiR-133aの発現を制御する転写因子であることが明らかにされた.そこで,染色およびウェスタンブロット法により高CO2条件におけるMEF2Dの発現について検討したところ,MEF2Dは気道平滑筋細胞の核の周辺に局在し,核におけるタンパク質量は減少していた.クロマチン免疫沈降法によりmiR-133a遺伝子のプロモーター領域へのMEF2Dの結合について検討したところ,高CO2条件においてMEF2Dの結合は減少していた.以上のことから,高CO2によりMEF2Dの核における局在が低下し,その結果,miR-133a遺伝子のプロモーター領域への結合が減少することによりmiR-133aの発現は抑制されると考えられた.
MEF2Dのウェスタンブロット法による解析において,タンパク質量の減少ともにタンパク質の断片化も観察された.これまでに,MEF2Dの断片化においてカスパーゼの関与が報告されていた.高CO2条件におけるカスパーゼによるMEF2Dの断片化について検討するため,カスパーゼの阻害剤を用いてMEF2Dの断片化およびmiR-133aの発現について検討したところ,高CO2によるMEF2Dの断片化およびmiR-133の発現の抑制は改善された.高CO2により活性化するカスパーゼを調べたところ,マウスの気道平滑筋細胞においてカスパーゼ7のみが高CO2により活性化されていた.カスパーゼ7をノックダウンしてMEF2Dの断片化およびmiR-133aの発現について検討したところ,高CO2によるMEF2Dの断片化およびmiR-133の発現の抑制は改善された.カスパーゼは細胞においてアポトーシスを制御することで知られている7).しかし,高CO2条件において気道平滑筋細胞のアポトーシスは認められなかった.以上のことから,高CO2はアポトーシスの制御機構とは異なるカスパーゼ7の活性化を介して気道平滑筋細胞を収縮させると考えられた.
カスパーゼの活性はCa2+依存性のシステインプロテアーゼであるカルパインにより制御されることが報告されていた8).マウスの気道平滑筋細胞におけるカルパインの活性について検討したところ,高CO2により一時的な上昇が観察された.高CO2条件において,組織に普遍的に発現するカルパイン1およびカルパイン2をノックダウンして気道平滑筋細胞におけるカスパーゼ7の活性について検討したところ,カルパイン1のノックダウンによりカスパーゼ7の活性は抑制された.一方,高CO2はマウスの気道平滑筋細胞において細胞におけるCa2+の濃度を上昇させ,また,Ca2+のキレート剤は高CO2によるカルパインの活性の上昇を抑制した.以上のことから,高CO2は気道平滑筋細胞におけるCa2+の濃度を上昇させ,カルパインの活性を介してカスパーゼ7を活性化させると考えられた(図1).
3.高CO2に曝露されたマウスにおいて気道の抵抗が上昇する
in vitroの実験系において認められた高CO2による気道平滑筋細胞の収縮の機序が,マウスのin vivoのモデルにおいても確認されるか検討した.高CO2かつ正常なO2に曝露された野生型のマウスの気道の収縮および気道の抵抗について検討したところ,気道の収縮はCO2の曝露の期間と比例して上昇し,また,気道の抵抗も上昇した.また,高CO2に曝露されたマウスの肺組織においては,miR-133aの発現は低下,RhoAの発現は上昇,ミオシン軽鎖はリン酸化されていた.一方,カスパーゼ7のノックアウトマウスにおいては,高CO2による気道の収縮および気道の抵抗の上昇は抑制され,また,高CO2によりひき起こされるmiR-133a-RhoA-ROCK-ミオシン軽鎖ホスファターゼ-ミオシン軽鎖というシグナル伝達経路の下流のシグナルも抑制されていた.これに対し,経気道的にmiR-133aの阻害剤を導入したカスパーゼ7のノックアウトマウスにおいては,野生型のマウスと同様に,高CO2により気道が収縮し,高CO2によりひき起こされる下流のシグナルも認められた.
4.高CO2血症を呈する慢性閉塞性肺疾患の患者において気道および呼吸の抵抗が上昇する
高CO2がヒトにおいても気道の収縮に影響をおよぼすのかどうか検討するため,慢性閉塞性肺疾患のコホート研究を用いて,高CO2血症を呈する重症の患者および高CO2血症を呈さない重症の患者を対象に,気道および呼吸の抵抗を測定した.その結果,高CO2血症をともなう患者においては気道および呼吸の抵抗が上昇し,また,非侵襲的な陽圧換気療法による介入により高CO2血症が改善された症例においては呼吸の抵抗が低下した.
おわりに
この研究において,筆者らは,CO2がシグナル伝達分子として気道平滑筋細胞の収縮に影響をおよぼすことを明らかにした.高CO2は気道平滑筋に有害であることが示唆されたが,高CO2血症を呈する慢性閉塞性肺疾患の患者において,高CO2を容認するか否かについては議論がある.その背景のひとつに,急性肺障害/急性呼吸窮迫症候群の患者においては,呼吸の管理において高CO2血症を容認するという概念が広く普及していることがある9).これは,肺の過膨張による正常な肺の障害を保護する目的で推奨された呼吸の管理法であるが,一方で,高CO2血症にともなう呼吸性のアシドーシスが抗炎症応答にも寄与することが示唆されており,このことから,治療のための高CO2血症という治療戦略も提唱されている.近年になり,慢性閉塞性肺疾患の患者において高CO2血症への介入が検討され3,4),その有効性が議論されている.この研究の成果は,これらの臨床研究を分子生物学的な観点からサポートする知見であり意義深い.
文 献
- Shigemura, M., Lecuona, E. & Sznajder, J. I.: Effects of hypercapnia on the lung. J. Physiol., 595, 2431-2437 (2017)[PubMed]
- Connors, A. F. Jr., Dawson, N. V., Thomas, C. et al.: Outcomes following acute exacerbation of severe chronic obstructive lung disease. The SUPPORT investigators (Study to Understand Prognoses and Preferences for Outcomes and Risks of Treatments). Am. J. Respir. Crit. Care Med., 154, 959-967 (1996)[PubMed]
- Murphy, P. B., Rehal, S., Arbane, G. et al.: Effect of home noninvasive ventilation with oxygen therapy vs oxygen therapy alone on hospital readmission or death after an acute COPD exacerbation: a eandomized clinical trial. JAMA, 317, 2177-2186 (2017)[PubMed]
- Kohnlein, T., Windisch, W., Kohler, D. et al.: Non-invasive positive pressure ventilation for the treatment of severe stable chronic obstructive pulmonary disease: a prospective, multicentre, randomised, controlled clinical trial. Lancet Respir. Med., 2, 698-705 (2014)[PubMed]
- Murthy, K. S.: Signaling for contraction and relaxation in smooth muscle of the gut. Annu. Rev. Physiol., 68, 345-374 (2006)[PubMed]
- Chiba, Y., Tanabe, M., Goto, K. et al.: Down-regulation of miR-133a contributes to up-regulation of RhoA in bronchial smooth muscle cells. Am. J. Respir. Crit. Care Med., 180, 713-719 (2009)[PubMed]
- Crawford, E. D. & Wells, J. A.: Caspase substrates and cellular remodeling. Annu. Rev. Biochem., 80, 1055-1087 (2011)[PubMed]
- Orrenius, S., Zhivotovsky, B. & Nicotera, P.: Regulation of cell death: the calcium-apoptosis link. Nat. Rev. Mol. Cell Biol., 4, 552-565 (2003)[PubMed]
- Acute Respiratory Distress Syndrome Network: Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N. Engl. J. Med., 342, 1301-1308 (2000)[PubMed]
活用したデータベースにかかわるキーワードと統合TVへのリンク
著者プロフィール
略歴:2011年 北海道大学大学院医学研究科博士課程 修了,2013年より米国Northwestern大学Feinberg School of Medicine博士研究員.
研究テーマ:CO2による生態制御の生物学.
関心事:生物の代謝機構におけるCO2の役割およびセンサー分子の探索.
Jacob I. Sznajder
米国Northwestern大学Feinberg School of Medicine教授.
研究室URL:https://labs.feinberg.northwestern.edu/sznajder/
© 2018 重村雅彦・Jacob I. Sznajder Licensed under CC 表示 2.1 日本
