ミトコンドリアの分裂と融合の停止が示したマイトファジーをひき起こすユビキチン化および非アルコール性脂肪性肝疾患の症状の抑制
山田達也・飯島美帆・瀬崎博美
(米国Johns Hopkins大学School of Medicine,Department of Cell Biology)
email:山田達也
DOI: 10.7875/first.author.2018.074
Mitochondrial stasis reveals p62-mediated ubiquitination in parkin-independent mitophagy and mitigates nonalcoholic fatty liver disease.
Tatsuya Yamada, Daisuke Murata, Yoshihiro Adachi, Kie Itoh, Shoichiro Kameoka, Atsushi Igarashi, Takashi Kato, Yoichi Araki, Richard L. Huganir, Ted M. Dawson, Toru Yanagawa, Koji Okamoto, Miho Iijima, Hiromi Sesaki
Cell Metabolism, 28, 588-604.e5 (2018)
細胞におけるエネルギーの工場であるミトコンドリアは好気呼吸によりATPの産生を担うと同時に,その副次的な影響としてつねに活性酸素にさらされる.ミトコンドリアはこの危険からのがれるため分裂と融合をダイナミックにくり返し恒常性を維持する.この研究において,筆者らは,ミトコンドリアの分裂の停止ではなく,ミトコンドリアの巨大化がマイトファジーとよばれるミトコンドリアの選択的なオートファジーの非効率化をひき起こし,組織の損傷の原因になることを明らかにした.さらに,このマイトファジーはp62-Keap1-Rbx1経路という新規の経路を介したユビキチン化によりひき起こされていた.同様のマイトファジーは非アルコール性脂肪性肝疾患のモデルマウスの肝臓においても阻害されており,それが肝臓の損傷をひき起こす原因のひとつとなっていた.非アルコール性脂肪性肝疾患におけるマイトファジーの阻害もミトコンドリアの巨大化が原因になっており,巨大化の抑制によりその症状は軽減された.この研究は,ミトコンドリアは細胞においてなぜダイナミックにふるまうのか,そして,そのダイナミクスの乱れがどのように疾患をひき起こすのかという疑問に答えた点で意義がある.
共生細菌を起源にもつミトコンドリアは分裂と融合をくり返す.分裂はミトコンドリアの細胞における分布およびターンオーバーと深くかかわり,融合はミトコンドリアの構成成分を混合しその不均一性を低下させると考えられてきた.ミトコンドリアの分裂と融合がどちらか一方にかたよると組織レベルおよび個体レベルにおいて重篤な損傷が生じることが明らかにされており1-3),ミトコンドリアの分裂あるいは融合にかかわる遺伝子の変異を原因とする疾患も特定されている4).しかしながら,なぜミトコンドリアが分裂と融合をくり返すのか? という本質的な問いに対する解答は得られていない.なぜなら,ミトコンドリアの分裂と融合のバランスがくずれると,同時に,巨大化あるいは断片化という大きさの変化も生じるためである.したがって,疾患をひき起こす原因はミトコンドリアの分裂と融合のバランスの破綻なのか,それとも,極端な巨大化あるいは断片化という大きさの変化が原因なのかは明確にされていなかった.この研究において,筆者らは,ミトコンドリアの分裂と融合が停止したものの,大きさは正常な状態と同等であるマウスモデルを作製し解析した.
in vivoにおいてミトコンドリアが分裂と融合をくり返す意義について明らかにするため,ダイナミン様GTPaseに由来するミトコンドリアの分裂と融合を阻害したin vivoモデルを作製した.ミトコンドリアに集積して分裂をひき起こすDrp1およびミトコンドリア内膜の融合に関与するOpa1に着目し,肝細胞に特異的なDrp1ノックアウトマウス,肝細胞に特異的なOpa1ノックアウトマウス,肝細胞に特異的なDrp1 Opa1ダブルノックアウトマウスを作製した.Drp1ノックアウトマウスの肝臓においてミトコンドリアは分裂が阻害されて巨大化し,Opa1ノックアウトマウスの肝臓においてミトコンドリアは融合の不全による断片化が生じた.Drp1 Opa1ダブルノックアウトマウスにおいてミトコンドリアの形態および大きさは野生型のマウスに類似しており,分裂と融合は阻害されているものの形態は正常な状態に近かった.これらのマウスモデルの肝臓の生理機能を解析したところ,Drp1ノックアウトマウスにおいては肝臓の損傷および血中の脂質の上昇,Opa1ノックアウトマウスにおいては血中の脂質の上昇とともに肝臓の萎縮がみられた.したがって,ミトコンドリアの分裂と融合がいずれかにかたよることにより肝臓の機能が低下することが確認された.一方,Drp1 Opa1ダブルノックアウトマウスの肝臓の機能は正常であった.したがって,ミトコンドリアの分裂と融合の平衡状態が破綻することによる肝臓の機能の障害は,強制的に平衡状態とすることにより軽減され,分裂と融合それ自体は必要ではなく形態を維持することが重要であることが明らかにされた.
DRP1遺伝子の欠損モデルにおいて,損傷の蓄積したミトコンドリアをオートファジーにより選択的に分解するマイトファジーに欠陥が生じることが知られている5,6).しかし,このマイトファジーの阻害が,ミトコンドリアの分裂の停止に起因するのか巨大化に起因するのかは不明であった.肝細胞に特異的なDrp1ノックアウトマウスの肝臓においてマイトファジーは阻害されていた.さらに,肝細胞に特異的なDrp1 Opa1ダブルノックアウトマウスの肝臓においてミトコンドリアの分裂は阻害されているのにもかかわらず,マイトファジーは正常に起こっていた.したがって,ミトコンドリアの形態や大きさを正常な状態に維持することがマイトファジーの遂行にとり重要であり,ミトコンドリアが分裂すること自体は必須ではないことが示唆された.さらに,マイトファジーの阻害が組織の損傷を起こす原因の一部であることも示唆された.
マイトファジーを起こすしくみとして,分解の対象となるミトコンドリアのユビキチン化,ユビキチン化されたミトコンドリアへのオートファジー受容体の結合,オートファジー受容体とLC3との結合によるオートファゴソームの動員,という過程がある.マイトファジーについてもっとも研究されているのがPink1-Parkin経路を介したミトコンドリア外膜のユビキチン化である7).Pink1はミトコンドリア内膜の膜電位が低下するとミトコンドリア外膜にとどまりユビキチンおよびParkinをリン酸化する.ユビキチンリガーゼであるParkinはリン酸化により活性化され標的をユビキチン化する8).しかしながら,Drp1遺伝子の欠損モデルのニューロンや心筋細胞においてPARKIN遺伝子を欠損させてもミトコンドリアのユビキチン化は消失しないことが示されている.肝細胞に特異的なDrp1ノックアウトマウスの肝臓においてマイトファジーは阻害されており,その結果として,ミトコンドリアにはユビキチン,オートファジー受容体であるp62,LC3が蓄積することが確認された.さらに,肝細胞に特異的なDrp1 Parkinダブルノックアウトマウスにおいても,これらのマイトファジーの中間体の蓄積は消失しなかった.したがって,in vivoにおいては,Parkinに非依存的な経路によりマイトファジーがひき起こされることが強く示唆された.
肝細胞に特異的なDrp1 p62ダブルノックアウトマウスの肝臓においてミトコンドリアに蓄積するLC3がいちじるしく減少したことから,LC3およびオートファゴソームのミトコンドリアへの動員にはp62が必須であることが示された.Drp1 p62ダブルノックアウトマウスの肝臓においてはユビキチンの蓄積も顕著に減少した.これらの結果から,ミトコンドリアはp62に依存的にユビキチン化されることが強く示唆され,ユビキチン化の下流においてはたらくと考えられていたp62は,ミトコンドリアのユビキチン化を促進する役割もはたすことが推察された.
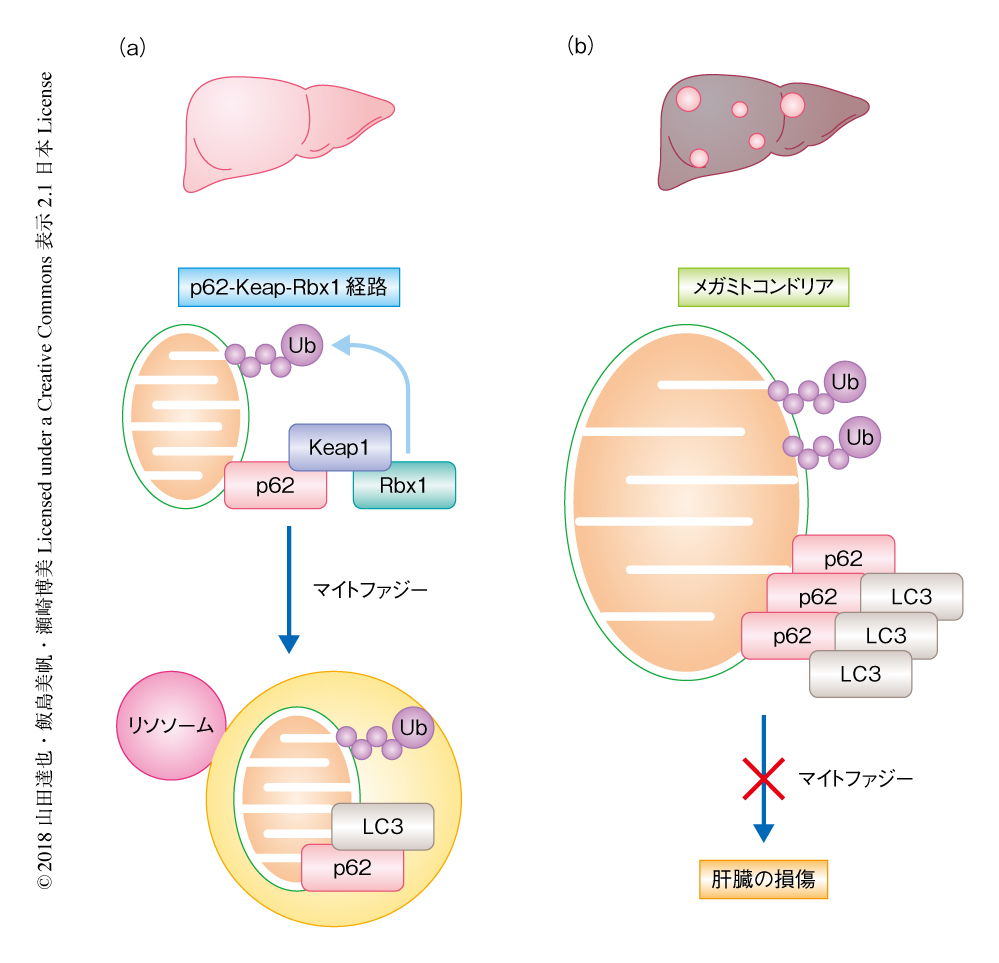
p62はKeap1結合ドメインをもつ.Keap1はCullin-Ringユビキチンリガーゼ複合体の構成タンパク質のひとつであることから,p62に依存的なミトコンドリアのユビキチン化はKeap1を含むユビキチンリガーゼ複合体が担うのではないかと仮説をたてた.これを検証した結果,ユビキチンリガーゼ複合体を形成するKeap1およびRbx1がp62に依存的にミトコンドリアに動員されユビキチン化を担うことが明らかにされた.したがって,p62はユビキチンを認識するオートファジー受容体のひとつであると同時に,ミトコンドリアのユビキチン化を促進する役割もはたしており,肝臓においてはp62-Keap1-Rbx1経路によるユビキチン化の促進がマイトファジーに寄与することが明らかにされた.
ミトコンドリアの形態を制御する遺伝子やマイトファジーにかかわる遺伝子の変異は複数の疾患にかかわることが明らかにされている4).ミトコンドリア外膜の融合を担うMFN2遺伝子の変異はCharcot-Marie-Tooth病2型,ミトコンドリア内膜の融合を担うOPA1遺伝子の変異は視神経の萎縮の原因になっており,マイトファジーにかかわるPARKIN遺伝子およびPINK1遺伝子の変異はパーキンソン病の患者に多いことが知られている.また,非アルコール性脂肪性肝疾患の患者の肝臓においては極端に巨大化したメガミトコンドリアが形成される.しかしながら,このメガミトコンドリアが病態の発現にどのように寄与するかは不明であった.そこで,マウスに非アルコール性脂肪性肝疾患を発症させる食餌をあたえることにより病態を模擬したモデルマウスを作製した.モデルマウスの肝臓においてもメガミトコンドリアが形成され,メガミトコンドリアにはユビキチンおよびp62が蓄積し,マイトファジーが阻害されていた.さらに,Keap1もミトコンドリアに動員されていた.したがって,非アルコール性脂肪性肝疾患の肝臓においてはメガミトコンドリアの形成がp62-Keap1-Rbx1経路を介したマイトファジーの遂行を阻んでいることが明らかにされた.ミトコンドリアの融合が阻害された肝細胞に特異的なOpa1ノックアウトマウスに同様の食餌をあたえると,ミトコンドリアの巨大化は抑制されマイトファジーの中間体の蓄積は確認されなかった.さらに,野生型のマウスから作製した非アルコール性脂肪性肝疾患のモデルマウスにおいては肝臓の損傷が強くひき起こされたのに対し,Opa1ノックアウトマウスから作製したモデルマウスにおいては肝臓の損傷は顕著に抑制された.これらのことから,メガミトコンドリアの形成がマイトファジーの遂行を阻害することにより肝臓の損傷をひき起こし,非アルコール性脂肪性肝疾患の病態の発現の原因のひとつとなると考えられた.
この研究においては,以下の3つの点が新たに示された(図1).1)肝細胞におけるミトコンドリアの分裂と融合の意義はミトコンドリアの大きさを制御することであると考えられた.なぜなら,ダイナミン様GTPaseに由来するミトコンドリアの分裂と融合を完全に阻害したとしても組織の機能は維持され,分裂と融合が一方にかたよった際に生じた表現型が回復したためである.2)in vivoの肝細胞におけるマイトファジーはPink1-Parkin経路に非依存的であり,p62-Keap1-Rbx1経路によりひき起こされる.さらに,ミトコンドリアの巨大化によりマイトファジーの効率的な遂行は阻害され,その結果として,肝臓の損傷につながると考えられた.3)非アルコール性脂肪性肝疾患において観察されるメガミトコンドリアは効率的なマイトファジーの阻害をひき起こし,肝臓の損傷につながると考えられた.OPA1遺伝子の欠損によりメガミトコンドリアの形成を阻害すると肝臓の損傷は軽減された.この研究により,非アルコール性脂肪性肝疾患をひき起こす原因の一部,および,その病状を回復させる治療の戦略が示唆されたのではないかと期待している.さらに研究を発展させ,非アルコール性脂肪性肝疾患の治療や予防に役だつことを願っている.
略歴:2015年 金沢大学大学院自然科学研究科 修了,同年より米国Johns Hopkins大学School of Medicineにて博士研究員.
研究テーマ:哺乳類のin vivoにおけるミトコンドリアの役割.
抱負:ミトコンドリアが多細胞生物の進化の鍵となった理由を解き明かしたい.
飯島 美帆(Miho Iijima)
米国Johns Hopkins大学School of Medicine准教授.
研究室URL:http://cellbio.jhmi.edu/people/faculty/miho-iijima-phd
瀬崎 博美(Hiromi Sesaki)
米国Johns Hopkins大学School of Medicine教授.
研究室URL:http://cellbio.jhmi.edu/people/faculty/hiromi-sesaki-phd
© 2018 山田達也・飯島美帆・瀬崎博美 Licensed under CC 表示 2.1 日本
(米国Johns Hopkins大学School of Medicine,Department of Cell Biology)
email:山田達也
DOI: 10.7875/first.author.2018.074
Mitochondrial stasis reveals p62-mediated ubiquitination in parkin-independent mitophagy and mitigates nonalcoholic fatty liver disease.
Tatsuya Yamada, Daisuke Murata, Yoshihiro Adachi, Kie Itoh, Shoichiro Kameoka, Atsushi Igarashi, Takashi Kato, Yoichi Araki, Richard L. Huganir, Ted M. Dawson, Toru Yanagawa, Koji Okamoto, Miho Iijima, Hiromi Sesaki
Cell Metabolism, 28, 588-604.e5 (2018)
要 約
細胞におけるエネルギーの工場であるミトコンドリアは好気呼吸によりATPの産生を担うと同時に,その副次的な影響としてつねに活性酸素にさらされる.ミトコンドリアはこの危険からのがれるため分裂と融合をダイナミックにくり返し恒常性を維持する.この研究において,筆者らは,ミトコンドリアの分裂の停止ではなく,ミトコンドリアの巨大化がマイトファジーとよばれるミトコンドリアの選択的なオートファジーの非効率化をひき起こし,組織の損傷の原因になることを明らかにした.さらに,このマイトファジーはp62-Keap1-Rbx1経路という新規の経路を介したユビキチン化によりひき起こされていた.同様のマイトファジーは非アルコール性脂肪性肝疾患のモデルマウスの肝臓においても阻害されており,それが肝臓の損傷をひき起こす原因のひとつとなっていた.非アルコール性脂肪性肝疾患におけるマイトファジーの阻害もミトコンドリアの巨大化が原因になっており,巨大化の抑制によりその症状は軽減された.この研究は,ミトコンドリアは細胞においてなぜダイナミックにふるまうのか,そして,そのダイナミクスの乱れがどのように疾患をひき起こすのかという疑問に答えた点で意義がある.
はじめに
共生細菌を起源にもつミトコンドリアは分裂と融合をくり返す.分裂はミトコンドリアの細胞における分布およびターンオーバーと深くかかわり,融合はミトコンドリアの構成成分を混合しその不均一性を低下させると考えられてきた.ミトコンドリアの分裂と融合がどちらか一方にかたよると組織レベルおよび個体レベルにおいて重篤な損傷が生じることが明らかにされており1-3),ミトコンドリアの分裂あるいは融合にかかわる遺伝子の変異を原因とする疾患も特定されている4).しかしながら,なぜミトコンドリアが分裂と融合をくり返すのか? という本質的な問いに対する解答は得られていない.なぜなら,ミトコンドリアの分裂と融合のバランスがくずれると,同時に,巨大化あるいは断片化という大きさの変化も生じるためである.したがって,疾患をひき起こす原因はミトコンドリアの分裂と融合のバランスの破綻なのか,それとも,極端な巨大化あるいは断片化という大きさの変化が原因なのかは明確にされていなかった.この研究において,筆者らは,ミトコンドリアの分裂と融合が停止したものの,大きさは正常な状態と同等であるマウスモデルを作製し解析した.
1.ミトコンドリアの分裂と融合のバランスが均衡することが重要である
in vivoにおいてミトコンドリアが分裂と融合をくり返す意義について明らかにするため,ダイナミン様GTPaseに由来するミトコンドリアの分裂と融合を阻害したin vivoモデルを作製した.ミトコンドリアに集積して分裂をひき起こすDrp1およびミトコンドリア内膜の融合に関与するOpa1に着目し,肝細胞に特異的なDrp1ノックアウトマウス,肝細胞に特異的なOpa1ノックアウトマウス,肝細胞に特異的なDrp1 Opa1ダブルノックアウトマウスを作製した.Drp1ノックアウトマウスの肝臓においてミトコンドリアは分裂が阻害されて巨大化し,Opa1ノックアウトマウスの肝臓においてミトコンドリアは融合の不全による断片化が生じた.Drp1 Opa1ダブルノックアウトマウスにおいてミトコンドリアの形態および大きさは野生型のマウスに類似しており,分裂と融合は阻害されているものの形態は正常な状態に近かった.これらのマウスモデルの肝臓の生理機能を解析したところ,Drp1ノックアウトマウスにおいては肝臓の損傷および血中の脂質の上昇,Opa1ノックアウトマウスにおいては血中の脂質の上昇とともに肝臓の萎縮がみられた.したがって,ミトコンドリアの分裂と融合がいずれかにかたよることにより肝臓の機能が低下することが確認された.一方,Drp1 Opa1ダブルノックアウトマウスの肝臓の機能は正常であった.したがって,ミトコンドリアの分裂と融合の平衡状態が破綻することによる肝臓の機能の障害は,強制的に平衡状態とすることにより軽減され,分裂と融合それ自体は必要ではなく形態を維持することが重要であることが明らかにされた.
DRP1遺伝子の欠損モデルにおいて,損傷の蓄積したミトコンドリアをオートファジーにより選択的に分解するマイトファジーに欠陥が生じることが知られている5,6).しかし,このマイトファジーの阻害が,ミトコンドリアの分裂の停止に起因するのか巨大化に起因するのかは不明であった.肝細胞に特異的なDrp1ノックアウトマウスの肝臓においてマイトファジーは阻害されていた.さらに,肝細胞に特異的なDrp1 Opa1ダブルノックアウトマウスの肝臓においてミトコンドリアの分裂は阻害されているのにもかかわらず,マイトファジーは正常に起こっていた.したがって,ミトコンドリアの形態や大きさを正常な状態に維持することがマイトファジーの遂行にとり重要であり,ミトコンドリアが分裂すること自体は必須ではないことが示唆された.さらに,マイトファジーの阻害が組織の損傷を起こす原因の一部であることも示唆された.
2.Parkinに非依存的なp62-Keap1-Rbx1経路を介した新規のマイトファジーの同定
マイトファジーを起こすしくみとして,分解の対象となるミトコンドリアのユビキチン化,ユビキチン化されたミトコンドリアへのオートファジー受容体の結合,オートファジー受容体とLC3との結合によるオートファゴソームの動員,という過程がある.マイトファジーについてもっとも研究されているのがPink1-Parkin経路を介したミトコンドリア外膜のユビキチン化である7).Pink1はミトコンドリア内膜の膜電位が低下するとミトコンドリア外膜にとどまりユビキチンおよびParkinをリン酸化する.ユビキチンリガーゼであるParkinはリン酸化により活性化され標的をユビキチン化する8).しかしながら,Drp1遺伝子の欠損モデルのニューロンや心筋細胞においてPARKIN遺伝子を欠損させてもミトコンドリアのユビキチン化は消失しないことが示されている.肝細胞に特異的なDrp1ノックアウトマウスの肝臓においてマイトファジーは阻害されており,その結果として,ミトコンドリアにはユビキチン,オートファジー受容体であるp62,LC3が蓄積することが確認された.さらに,肝細胞に特異的なDrp1 Parkinダブルノックアウトマウスにおいても,これらのマイトファジーの中間体の蓄積は消失しなかった.したがって,in vivoにおいては,Parkinに非依存的な経路によりマイトファジーがひき起こされることが強く示唆された.
肝細胞に特異的なDrp1 p62ダブルノックアウトマウスの肝臓においてミトコンドリアに蓄積するLC3がいちじるしく減少したことから,LC3およびオートファゴソームのミトコンドリアへの動員にはp62が必須であることが示された.Drp1 p62ダブルノックアウトマウスの肝臓においてはユビキチンの蓄積も顕著に減少した.これらの結果から,ミトコンドリアはp62に依存的にユビキチン化されることが強く示唆され,ユビキチン化の下流においてはたらくと考えられていたp62は,ミトコンドリアのユビキチン化を促進する役割もはたすことが推察された.
p62はKeap1結合ドメインをもつ.Keap1はCullin-Ringユビキチンリガーゼ複合体の構成タンパク質のひとつであることから,p62に依存的なミトコンドリアのユビキチン化はKeap1を含むユビキチンリガーゼ複合体が担うのではないかと仮説をたてた.これを検証した結果,ユビキチンリガーゼ複合体を形成するKeap1およびRbx1がp62に依存的にミトコンドリアに動員されユビキチン化を担うことが明らかにされた.したがって,p62はユビキチンを認識するオートファジー受容体のひとつであると同時に,ミトコンドリアのユビキチン化を促進する役割もはたしており,肝臓においてはp62-Keap1-Rbx1経路によるユビキチン化の促進がマイトファジーに寄与することが明らかにされた.
3.非アルコール性脂肪性肝疾患モデルの病態はメガミトコンドリアの形成の抑制により軽減される
ミトコンドリアの形態を制御する遺伝子やマイトファジーにかかわる遺伝子の変異は複数の疾患にかかわることが明らかにされている4).ミトコンドリア外膜の融合を担うMFN2遺伝子の変異はCharcot-Marie-Tooth病2型,ミトコンドリア内膜の融合を担うOPA1遺伝子の変異は視神経の萎縮の原因になっており,マイトファジーにかかわるPARKIN遺伝子およびPINK1遺伝子の変異はパーキンソン病の患者に多いことが知られている.また,非アルコール性脂肪性肝疾患の患者の肝臓においては極端に巨大化したメガミトコンドリアが形成される.しかしながら,このメガミトコンドリアが病態の発現にどのように寄与するかは不明であった.そこで,マウスに非アルコール性脂肪性肝疾患を発症させる食餌をあたえることにより病態を模擬したモデルマウスを作製した.モデルマウスの肝臓においてもメガミトコンドリアが形成され,メガミトコンドリアにはユビキチンおよびp62が蓄積し,マイトファジーが阻害されていた.さらに,Keap1もミトコンドリアに動員されていた.したがって,非アルコール性脂肪性肝疾患の肝臓においてはメガミトコンドリアの形成がp62-Keap1-Rbx1経路を介したマイトファジーの遂行を阻んでいることが明らかにされた.ミトコンドリアの融合が阻害された肝細胞に特異的なOpa1ノックアウトマウスに同様の食餌をあたえると,ミトコンドリアの巨大化は抑制されマイトファジーの中間体の蓄積は確認されなかった.さらに,野生型のマウスから作製した非アルコール性脂肪性肝疾患のモデルマウスにおいては肝臓の損傷が強くひき起こされたのに対し,Opa1ノックアウトマウスから作製したモデルマウスにおいては肝臓の損傷は顕著に抑制された.これらのことから,メガミトコンドリアの形成がマイトファジーの遂行を阻害することにより肝臓の損傷をひき起こし,非アルコール性脂肪性肝疾患の病態の発現の原因のひとつとなると考えられた.
おわりに
この研究においては,以下の3つの点が新たに示された(図1).1)肝細胞におけるミトコンドリアの分裂と融合の意義はミトコンドリアの大きさを制御することであると考えられた.なぜなら,ダイナミン様GTPaseに由来するミトコンドリアの分裂と融合を完全に阻害したとしても組織の機能は維持され,分裂と融合が一方にかたよった際に生じた表現型が回復したためである.2)in vivoの肝細胞におけるマイトファジーはPink1-Parkin経路に非依存的であり,p62-Keap1-Rbx1経路によりひき起こされる.さらに,ミトコンドリアの巨大化によりマイトファジーの効率的な遂行は阻害され,その結果として,肝臓の損傷につながると考えられた.3)非アルコール性脂肪性肝疾患において観察されるメガミトコンドリアは効率的なマイトファジーの阻害をひき起こし,肝臓の損傷につながると考えられた.OPA1遺伝子の欠損によりメガミトコンドリアの形成を阻害すると肝臓の損傷は軽減された.この研究により,非アルコール性脂肪性肝疾患をひき起こす原因の一部,および,その病状を回復させる治療の戦略が示唆されたのではないかと期待している.さらに研究を発展させ,非アルコール性脂肪性肝疾患の治療や予防に役だつことを願っている.
文 献
- Wakabayashi, J., Zhang, Z., Wakabayashi, N. et al.: The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol., 186, 805-816 (2009)[PubMed]
- Chen, H., Vermulst, M., Wang, Y. E. et al.: Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell, 141, 280-289 (2010)[PubMed]
- Tezze, C., Romanello, V., Desbats, M. A. et al.: Age-associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab., 25, 1374-1389 (2017)[PubMed]
- Archer, S. L.: Mitochondrial dynamics: mitochondrial fission and fusion in human diseases. N. Engl. J. Med., 369, 2236-2251 (2013)[PubMed]
- Kageyama, Y., Zhang, Z., Roda, R. et al.: Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J. Cell Biol., 197, 535-551(2012)[PubMed]
- Kageyama, Y., Hoshijima, M., Seo, K. et al.: Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J., 33, 2798-2813 (2014)[PubMed]
- Pickels, S., Vigie, P. & Youle, R. J.: Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol., 28, R170-R185 (2018)[PubMed]
- Wauer, T., Simicek, M., Schubert, A. et al.: Mechanism of phospho-ubiquitin-induced PARKIN activation. Nature, 524, 370-374 (2015)[PubMed]
活用したデータベースにかかわるキーワードと統合TVへのリンク
著者プロフィール
略歴:2015年 金沢大学大学院自然科学研究科 修了,同年より米国Johns Hopkins大学School of Medicineにて博士研究員.
研究テーマ:哺乳類のin vivoにおけるミトコンドリアの役割.
抱負:ミトコンドリアが多細胞生物の進化の鍵となった理由を解き明かしたい.
飯島 美帆(Miho Iijima)
米国Johns Hopkins大学School of Medicine准教授.
研究室URL:http://cellbio.jhmi.edu/people/faculty/miho-iijima-phd
瀬崎 博美(Hiromi Sesaki)
米国Johns Hopkins大学School of Medicine教授.
研究室URL:http://cellbio.jhmi.edu/people/faculty/hiromi-sesaki-phd
© 2018 山田達也・飯島美帆・瀬崎博美 Licensed under CC 表示 2.1 日本
