個体を用いた新しいがん治療薬の創薬の基盤
園下将大1・Ross L. Cagan 1・Arvin C. Dar 2
(米国Icahn School of Medicine at Mount Sinai,1 Department of Cell, Developmental and Regenerative Biology,2 Department of Oncological Sciences)
email:園下将大
DOI: 10.7875/first.author.2018.017
A whole-animal platform to advance a clinical kinase inhibitor into new disease space.
Masahiro Sonoshita, Alex P. Scopton, Peter M. U. Ung, Matthew A. Murray, Lisa Silber, Andres Y. Maldonado, Alexander Real, Avner Schlessinger, Ross L. Cagan, Arvin C. Dar
Nature Chemical Biology, 14, 291-298 (2018)
既存薬の化学構造の改変はがん治療薬の迅速な開発や適応の拡大において魅力的な戦略のひとつである.しかし,多くの場合,高い効果かつ低い毒性をもたらす標的の阻害プロファイルが同定できないため,その実現は容易ではない.この研究において,筆者らは,阻害プロファイルの同定およびその情報にもとづく既存薬の化学構造の最適化による新しい創薬の手法を開発した.例として,キナーゼを阻害する分子標的薬であるソラフェニブと,米国においてソラフェニブが未適応である甲状腺髄様がんを対象として,化学遺伝学的なスクリーニングとin silicoモデリング解析とを融合することにより,ショウジョウバエの甲状腺髄様がんモデルにおいてソラフェニブの抗腫瘍効果を増強させるキナーゼおよび減弱させるキナーゼを同定した.抗腫瘍効果を減弱させるキナーゼとして同定されたRAFあるいはMKNKと結合することにより,ソラフェニブの抗腫瘍効果は著明に低下した.そこで,これらのキナーゼに結合しなくなるようソラフェニブの化学構造を改変することにより,前臨床甲状腺髄様がんモデルにおいてソラフェニブよりはるかに高い抗腫瘍効果を示す新規の化合物が創出された.これらの結果から,既存薬の多重薬理学的な阻害プロファイルの最適化が高い効果かつ低い毒性をもつリード化合物の創出に有効であり,この手法がそのための論理的な基盤になることが示された.
キナーゼは生体の恒常性の維持に重要な役割をはたすが,過剰な発現や活性化型の変異によりがんをはじめとする種々の疾患をひき起こす.現在まで,米国食品医薬品局によりこれらの疾患に対し37種類のキナーゼ阻害薬が分子標的薬として認可されている.近年のゲノム解析技術の進歩にともない,変異型のキナーゼがさまざまな種類のがんに対する治療の有望な標的になると期待されている.しかし,実際にがんにおいて変異の同定されたキナーゼは依然として少数にとどまるだけでなく,治験や臨床を問わずキナーゼ阻害薬は高い毒性を示すなど,ゲノム情報の治療への活用は少数の例外を除き依然として容易ではない.実際,新規のがん治療薬の認可率は約7%にとどまり,主要な疾患のなかでは最低となっている1).
キナーゼ阻害薬をはじめとする分子標的薬が抗腫瘍効果を発揮するために,がんの発生の原因となるタンパク質の阻害はしばしば必須であるが,単独の標的の阻害だけでは十分ではないことが多い2).実際,本来の標的にくわえ,オフターゲットに対する阻害も薬効を左右することが示唆されている3).この研究において,筆者らは,迅速な創薬を達成するため,キナーゼ阻害薬を対象として既存薬の化学構造を合理的に改変し本来の標的とオフターゲットの阻害のバランスを最適化する多重薬理学的な手法の開発に取り組んだ.米国食品医薬品局により認可されているキナーゼ阻害薬は,生物学的な利用能,体内分布,代謝などの特性がヒトにおいて良好であることが確認されている.そこで,これらの特性を維持しつつ抗腫瘍効果を増強させることにより,これまでほとんど存在しなかったリード化合物の創出のための明確な基盤を確立することをめざした.
甲状腺髄様がんは受容体チロシンキナーゼであるRETが活性化型の変異を獲得することにより発症する.以前に,筆者らの研究グループは,ショウジョウバエにおいてptc遺伝子のプロモーターにより変異型のRetの発現が促進される甲状腺髄様がんモデルについて報告した4).このショウジョウバエは25℃で培養すると体内に腫瘍様の病変が発生し成虫になるまでにすべての個体が死亡する.筆者らの研究グループは,このショウジョウバエの甲状腺髄様がんモデルを使用し,Retによりもたらされる致死性を低下させる化合物,とくに,複数のキナーゼを阻害する多重薬理学的なキナーゼ阻害薬の化学遺伝学的なスクリーニングを実施してきた4,5).
ショウジョウバエの甲状腺髄様がんモデルに米国食品医薬品局により認可されているすべてのキナーゼ阻害薬(2016年の時点で,31種類)を経口投与し抗腫瘍効果を比較した.その結果,ソラフェニブ,レゴラフェニブ,トラメチニブの3つが生存率を弱く上昇させることがわかった.ソラフェニブとレゴラフェニブは類縁体でありRETやRAFをはじめとする複数のキナーゼを阻害する.一方,トラメチニブはMEK1およびMEK2の選択的な阻害薬である.これらの結果から,RET-MAPKシグナル伝達経路の阻害が甲状腺髄様がんの抑制に重要であることが示唆された.
米国において,ソラフェニブは肝臓がんや腎臓がんなどに対し認可されている.甲状腺髄様がんに対しては未認可であるが,ヒトの甲状腺髄様がん細胞の増殖を抑制することが報告されている6).治験においても甲状腺髄様がんに抗腫瘍効果を発揮する場合があるが,患者の死亡を含む重篤な副作用が知られており,肝臓がんや腎臓がんの患者でも下痢や皮膚がんを誘発する7).実際に,ショウジョウバエの甲状腺髄様がんモデルにおいてもソラフェニブには毒性が観察され,生存率も5%程度と低く,大きな改善の余地があった.そこで,ソラフェニブを対象として,RETにより発生が駆動される甲状腺髄様がんに対し高い抗腫瘍効果を得ることを目的とした化学構造の最適化のための合理的な手法の開発に取り組んだ.
ソラフェニブの化学構造を,ヒンジバインダー,スペーサー,リンカー,キャップ,の4つのユニットに便宜的に分割した.そして,おのおのの派生体を合成し,それぞれを組み合わせて約100種類のソラフェニブの類縁体を合成した.これらをショウジョウバエの甲状腺髄様がんモデルに投与したところ,そのうちTCI-4がソラフェニブよりも高い抗腫瘍効果を示すことがわかった.TCI-4はソラフェニブと比較して,F原子を付加しCl原子を除去したキャップをもつが,そのほかはソラフェニブと同一であった.キャップは,キナーゼの活性の制御に重要なAsp-Phe-Glyポケットと結合する領域である.このようなソラフェニブの類縁体の構造活性相関の解析により,キャップを改変することで抗腫瘍効果を大きく増強させられることが示唆された(図1).
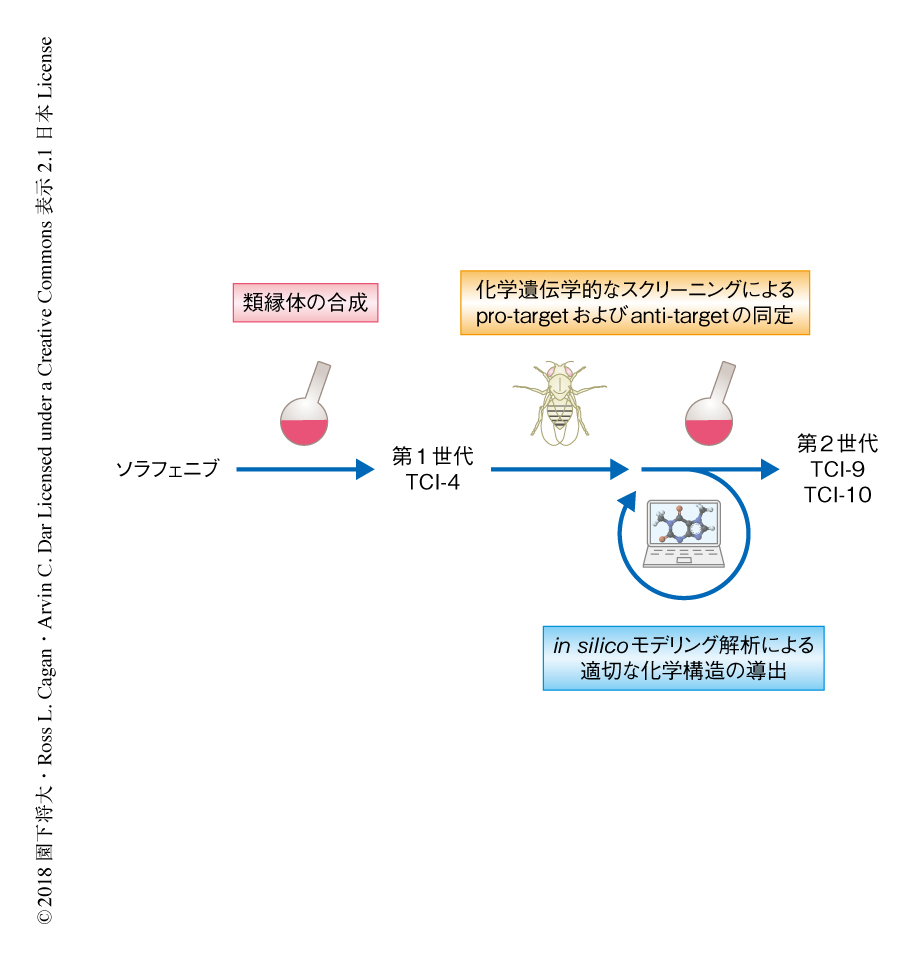
TCI-4の抗腫瘍効果をさらに増強させる方法について検討した.ショウジョウバエを用いる利点のひとつに,注目する表現型に影響をおよぼす遺伝子を容易に同定できることがあげられる5).ショウジョウバエのすべてのキナーゼ遺伝子について,おのおののヘテロ接合性変異をショウジョウバエの甲状腺髄様がんモデルに導入してTCI-4を投与することにより,TCI-4の抗腫瘍効果に影響をおよぼす遺伝子を化学遺伝学的にスクリーニングした.その結果,Eph遺伝子やSrc42A遺伝子のヘテロ接合性変異によりTCI-4の抗腫瘍効果はいちじるしく増強された一方,Lk6遺伝子のヘテロ接合性変異によりTCI-4の抗腫瘍効果は大きく減弱された.抗腫瘍効果を大きく増強させるものを“pro-target”,抗腫瘍効果を減弱させるものを“anti-target”と名づけ,pro-targetとして22遺伝子,anti-targetとして8遺伝子を同定した(図1).これらの化学遺伝学的なスクリーニングにより,抗腫瘍効果の増強のためにTCI-4が阻害すべきあるいはすべきでない遺伝子の情報が得られた.
in vitroにおける結合実験により,TCI-4はanti-targetとして同定されたMKNKおよびBRAF(ショウジョウバエのLk6およびPhlのヒトにおけるホモログ)と結合することが確認された.そこで,このTCI-4とanti-targetとの結合がTCI-4の抗腫瘍効果を減弱させると考え,この結合が起こらないようにTCI-4の化学構造を改変すれば抗腫瘍効果が増強されると考えた.
TCI-4を中心に実施した構造活性相関の解析の結果,キャップに存在するCF3基のF原子の数を減らすと抗腫瘍効果が減弱されることがわかった.このことから,CF3基が抗腫瘍効果を規定する重要な修飾基であることが示された.また,in silicoモデリング解析8) により,キャップが結合するAsp-Phe-Glyポケットの体積をpro-targetであるRETとanti-targetであるMKNKおよびBRAFとのあいだで比較したところ,RETよりもMKNKおよびBRAFのほうがAsp-Phe-Glyポケットは小さいことがわかった.そこで,RETのもつAsp-Phe-Glyポケットとの結合能は維持しつつも,MKNKあるいはBRAFのもつAsp-Phe-Glyポケットとの結合能を低下させるために,キャップにCF3基よりやや大きいC2F5をもつTCI-9を合成した(図1).TCI-9をショウジョウバエの甲状腺髄様がんモデルに投与したところ,TCI-9はTCI-4よりも高い抗腫瘍効果を示した.そこで,さらに大きな修飾基であるiso-C3F7をもつTCI-10を合成した(図1).TCI-10をショウジョウバエの甲状腺髄様がんモデルに投与したところ,84%の生存率という顕著な抗腫瘍効果を示した.これは,現在までに筆者らがショウジョウバエの甲状腺髄様がんモデルへの投与により得たなかでもっとも高いものである.
実際に,TCI-9およびTCI-10はソラフェニブに比べBRAFおよびMKNKとの結合能が低下していた.MKNKはERKによりリン酸化されることで活性化し,RAS-MAPKシグナル伝達経路を阻害する負のフィードバックループを駆動することが示唆されている9).さらに,TCI-4はMKNKと結合してその活性を阻害することによりこの負のフィードバックループを抑制するためRAS-MAPKシグナル伝達経路の活性が維持されること,一方,TCI-9およびTCI-10はMKNKとの結合能が低下しているためMKNKの活性が高く維持され,この負のフィードバックループが機能することによりRAS-MAPKシグナル伝達経路が抑制されることも見い出された.
以上の結果から,化学遺伝学的なスクリーニングおよびin silicoモデリング解析から明らかにされたソラフェニブの類縁体におけるキャップの大型化がショウジョウバエの甲状腺髄様がんモデルにおける抗腫瘍効果の増強に直結すると結論された.
甲状腺髄様がんに対するTCI-10の抗腫瘍効果についてさらに検討するため,ヒトの甲状腺髄様がんに由来するTT細胞をヌードマウスの皮下に移植して腫瘍を形成させ,TCI-10,ソラフェニブ,現在の甲状腺髄様がんの標準的な治療薬であるカボザンチニブを投与した.その結果,TCI-10は投与されたマウスの75%に部分寛解や完全寛解をもたらした.この抗腫瘍効果は,寛解をまったくもたらさなかったソラフェニブやカボザンチニブよりもはるかに強力なものであった.また,TCI-10はマウスにおける副作用を発現しない最大投与量が一般の薬物よりも高く,30日間の投与によりマウスの体重や行動に影響をおよぼさなかった.これらの結果から,TCI-10は抗甲状腺髄様がんに対する抗腫瘍効果およびin vivoにおける好ましい動態を兼ね備えたリード化合物であることが示された.
この研究において,筆者らは,ショウジョウバエにおける化学遺伝学的なスクリーニングとin silicoモデリング解析とを融合し,認可薬から論理的に新規のリード化合物を創出する手法を開発した.おもに,ソラフェニブの類縁体の合成,anti-targetの同定,anti-targetと結合しない化学構造の導出,の3段階で甲状腺髄様がんの動物モデルにおいて高い抗腫瘍効果を示すリード化合物を創出した(図1).大きな特色として,ショウジョウバエを創薬の研究に活用することにより,遺伝学的な網羅的な解析および個体における迅速な薬効の評価が可能となった点,そして,異分野融合により効率的な基盤が確立された点をあげたい.
また,化学的な観点から,F原子による修飾が抗腫瘍効果の増強に有効であった点は興味深い.近年,F原子による既存薬の修飾は大きな注目をあつめているためである.実際,複数のF原子をもつ認可薬が増加してきており,7つのF原子をもつエンザルタミドやアプレピタントなどはその例である10).F原子の物性がどのように薬効に影響するかは完全には解明されていないものの,この研究において示されたC-F鎖による修飾は今後も化学構造の改変の有効な手段になる可能性がある.
これまで,がんの創薬の分野においては,単一の標的の阻害をめざす分子標的薬が大きな注目をあつめてきた.この研究において示された創薬の基盤はこれと相補的なものであり,低費用で効果的に新規のリード化合物を創出することが可能になると期待される.さらに,がんの発生を促進するキナーゼシグナル伝達ネットワークに対し多重薬理学的な化合物が有効であることも示された.この手法は,神経疾患や心疾患など創薬の困難な疾患にも適用できる可能性があり,創薬の分野を大きく変革する可能性をひめている.今後も,この手法の発展と応用をつうじ,がんをはじめとする疾患に苦しむ人たちの救済に貢献したい.
略歴:2004年 京都大学大学院医学研究科 修了,同 助教,同 講師,同 准教授を経て,2013年より米国Icahn School of Medicine at Mount Sinai研究員.
研究テーマ:がんの発生の機序の解明およびがんの治療薬の創出.
Ross L. Cagan
米国Icahn School of Medicine at Mount SinaiにてProfessor.
研究室URL:http://drb.mssm.edu/labs/cagan.html
Arvin C. Dar
米国Icahn School of Medicine at Mount SinaiにてAssistant Professor.
研究室URL:http://labs.icahn.mssm.edu/darlab/
© 2018 園下将大・Ross L. Cagan・Arvin C. Dar Licensed under CC 表示 2.1 日本
(米国Icahn School of Medicine at Mount Sinai,1 Department of Cell, Developmental and Regenerative Biology,2 Department of Oncological Sciences)
email:園下将大
DOI: 10.7875/first.author.2018.017
A whole-animal platform to advance a clinical kinase inhibitor into new disease space.
Masahiro Sonoshita, Alex P. Scopton, Peter M. U. Ung, Matthew A. Murray, Lisa Silber, Andres Y. Maldonado, Alexander Real, Avner Schlessinger, Ross L. Cagan, Arvin C. Dar
Nature Chemical Biology, 14, 291-298 (2018)
要 約
既存薬の化学構造の改変はがん治療薬の迅速な開発や適応の拡大において魅力的な戦略のひとつである.しかし,多くの場合,高い効果かつ低い毒性をもたらす標的の阻害プロファイルが同定できないため,その実現は容易ではない.この研究において,筆者らは,阻害プロファイルの同定およびその情報にもとづく既存薬の化学構造の最適化による新しい創薬の手法を開発した.例として,キナーゼを阻害する分子標的薬であるソラフェニブと,米国においてソラフェニブが未適応である甲状腺髄様がんを対象として,化学遺伝学的なスクリーニングとin silicoモデリング解析とを融合することにより,ショウジョウバエの甲状腺髄様がんモデルにおいてソラフェニブの抗腫瘍効果を増強させるキナーゼおよび減弱させるキナーゼを同定した.抗腫瘍効果を減弱させるキナーゼとして同定されたRAFあるいはMKNKと結合することにより,ソラフェニブの抗腫瘍効果は著明に低下した.そこで,これらのキナーゼに結合しなくなるようソラフェニブの化学構造を改変することにより,前臨床甲状腺髄様がんモデルにおいてソラフェニブよりはるかに高い抗腫瘍効果を示す新規の化合物が創出された.これらの結果から,既存薬の多重薬理学的な阻害プロファイルの最適化が高い効果かつ低い毒性をもつリード化合物の創出に有効であり,この手法がそのための論理的な基盤になることが示された.
はじめに
キナーゼは生体の恒常性の維持に重要な役割をはたすが,過剰な発現や活性化型の変異によりがんをはじめとする種々の疾患をひき起こす.現在まで,米国食品医薬品局によりこれらの疾患に対し37種類のキナーゼ阻害薬が分子標的薬として認可されている.近年のゲノム解析技術の進歩にともない,変異型のキナーゼがさまざまな種類のがんに対する治療の有望な標的になると期待されている.しかし,実際にがんにおいて変異の同定されたキナーゼは依然として少数にとどまるだけでなく,治験や臨床を問わずキナーゼ阻害薬は高い毒性を示すなど,ゲノム情報の治療への活用は少数の例外を除き依然として容易ではない.実際,新規のがん治療薬の認可率は約7%にとどまり,主要な疾患のなかでは最低となっている1).
キナーゼ阻害薬をはじめとする分子標的薬が抗腫瘍効果を発揮するために,がんの発生の原因となるタンパク質の阻害はしばしば必須であるが,単独の標的の阻害だけでは十分ではないことが多い2).実際,本来の標的にくわえ,オフターゲットに対する阻害も薬効を左右することが示唆されている3).この研究において,筆者らは,迅速な創薬を達成するため,キナーゼ阻害薬を対象として既存薬の化学構造を合理的に改変し本来の標的とオフターゲットの阻害のバランスを最適化する多重薬理学的な手法の開発に取り組んだ.米国食品医薬品局により認可されているキナーゼ阻害薬は,生物学的な利用能,体内分布,代謝などの特性がヒトにおいて良好であることが確認されている.そこで,これらの特性を維持しつつ抗腫瘍効果を増強させることにより,これまでほとんど存在しなかったリード化合物の創出のための明確な基盤を確立することをめざした.
1.ソラフェニブはショウジョウバエの甲状腺髄様がんモデルにおいて弱い抗腫瘍効果を発揮する
甲状腺髄様がんは受容体チロシンキナーゼであるRETが活性化型の変異を獲得することにより発症する.以前に,筆者らの研究グループは,ショウジョウバエにおいてptc遺伝子のプロモーターにより変異型のRetの発現が促進される甲状腺髄様がんモデルについて報告した4).このショウジョウバエは25℃で培養すると体内に腫瘍様の病変が発生し成虫になるまでにすべての個体が死亡する.筆者らの研究グループは,このショウジョウバエの甲状腺髄様がんモデルを使用し,Retによりもたらされる致死性を低下させる化合物,とくに,複数のキナーゼを阻害する多重薬理学的なキナーゼ阻害薬の化学遺伝学的なスクリーニングを実施してきた4,5).
ショウジョウバエの甲状腺髄様がんモデルに米国食品医薬品局により認可されているすべてのキナーゼ阻害薬(2016年の時点で,31種類)を経口投与し抗腫瘍効果を比較した.その結果,ソラフェニブ,レゴラフェニブ,トラメチニブの3つが生存率を弱く上昇させることがわかった.ソラフェニブとレゴラフェニブは類縁体でありRETやRAFをはじめとする複数のキナーゼを阻害する.一方,トラメチニブはMEK1およびMEK2の選択的な阻害薬である.これらの結果から,RET-MAPKシグナル伝達経路の阻害が甲状腺髄様がんの抑制に重要であることが示唆された.
米国において,ソラフェニブは肝臓がんや腎臓がんなどに対し認可されている.甲状腺髄様がんに対しては未認可であるが,ヒトの甲状腺髄様がん細胞の増殖を抑制することが報告されている6).治験においても甲状腺髄様がんに抗腫瘍効果を発揮する場合があるが,患者の死亡を含む重篤な副作用が知られており,肝臓がんや腎臓がんの患者でも下痢や皮膚がんを誘発する7).実際に,ショウジョウバエの甲状腺髄様がんモデルにおいてもソラフェニブには毒性が観察され,生存率も5%程度と低く,大きな改善の余地があった.そこで,ソラフェニブを対象として,RETにより発生が駆動される甲状腺髄様がんに対し高い抗腫瘍効果を得ることを目的とした化学構造の最適化のための合理的な手法の開発に取り組んだ.
2.ソラフェニブの類縁体の構造活性相関の解析
ソラフェニブの化学構造を,ヒンジバインダー,スペーサー,リンカー,キャップ,の4つのユニットに便宜的に分割した.そして,おのおのの派生体を合成し,それぞれを組み合わせて約100種類のソラフェニブの類縁体を合成した.これらをショウジョウバエの甲状腺髄様がんモデルに投与したところ,そのうちTCI-4がソラフェニブよりも高い抗腫瘍効果を示すことがわかった.TCI-4はソラフェニブと比較して,F原子を付加しCl原子を除去したキャップをもつが,そのほかはソラフェニブと同一であった.キャップは,キナーゼの活性の制御に重要なAsp-Phe-Glyポケットと結合する領域である.このようなソラフェニブの類縁体の構造活性相関の解析により,キャップを改変することで抗腫瘍効果を大きく増強させられることが示唆された(図1).
3.TCI-4のpro-targetおよびanti-targetの同定
TCI-4の抗腫瘍効果をさらに増強させる方法について検討した.ショウジョウバエを用いる利点のひとつに,注目する表現型に影響をおよぼす遺伝子を容易に同定できることがあげられる5).ショウジョウバエのすべてのキナーゼ遺伝子について,おのおののヘテロ接合性変異をショウジョウバエの甲状腺髄様がんモデルに導入してTCI-4を投与することにより,TCI-4の抗腫瘍効果に影響をおよぼす遺伝子を化学遺伝学的にスクリーニングした.その結果,Eph遺伝子やSrc42A遺伝子のヘテロ接合性変異によりTCI-4の抗腫瘍効果はいちじるしく増強された一方,Lk6遺伝子のヘテロ接合性変異によりTCI-4の抗腫瘍効果は大きく減弱された.抗腫瘍効果を大きく増強させるものを“pro-target”,抗腫瘍効果を減弱させるものを“anti-target”と名づけ,pro-targetとして22遺伝子,anti-targetとして8遺伝子を同定した(図1).これらの化学遺伝学的なスクリーニングにより,抗腫瘍効果の増強のためにTCI-4が阻害すべきあるいはすべきでない遺伝子の情報が得られた.
4.anti-targetであるMKNKの情報にもとづくTCI-9およびTCI-10の創出
in vitroにおける結合実験により,TCI-4はanti-targetとして同定されたMKNKおよびBRAF(ショウジョウバエのLk6およびPhlのヒトにおけるホモログ)と結合することが確認された.そこで,このTCI-4とanti-targetとの結合がTCI-4の抗腫瘍効果を減弱させると考え,この結合が起こらないようにTCI-4の化学構造を改変すれば抗腫瘍効果が増強されると考えた.
TCI-4を中心に実施した構造活性相関の解析の結果,キャップに存在するCF3基のF原子の数を減らすと抗腫瘍効果が減弱されることがわかった.このことから,CF3基が抗腫瘍効果を規定する重要な修飾基であることが示された.また,in silicoモデリング解析8) により,キャップが結合するAsp-Phe-Glyポケットの体積をpro-targetであるRETとanti-targetであるMKNKおよびBRAFとのあいだで比較したところ,RETよりもMKNKおよびBRAFのほうがAsp-Phe-Glyポケットは小さいことがわかった.そこで,RETのもつAsp-Phe-Glyポケットとの結合能は維持しつつも,MKNKあるいはBRAFのもつAsp-Phe-Glyポケットとの結合能を低下させるために,キャップにCF3基よりやや大きいC2F5をもつTCI-9を合成した(図1).TCI-9をショウジョウバエの甲状腺髄様がんモデルに投与したところ,TCI-9はTCI-4よりも高い抗腫瘍効果を示した.そこで,さらに大きな修飾基であるiso-C3F7をもつTCI-10を合成した(図1).TCI-10をショウジョウバエの甲状腺髄様がんモデルに投与したところ,84%の生存率という顕著な抗腫瘍効果を示した.これは,現在までに筆者らがショウジョウバエの甲状腺髄様がんモデルへの投与により得たなかでもっとも高いものである.
実際に,TCI-9およびTCI-10はソラフェニブに比べBRAFおよびMKNKとの結合能が低下していた.MKNKはERKによりリン酸化されることで活性化し,RAS-MAPKシグナル伝達経路を阻害する負のフィードバックループを駆動することが示唆されている9).さらに,TCI-4はMKNKと結合してその活性を阻害することによりこの負のフィードバックループを抑制するためRAS-MAPKシグナル伝達経路の活性が維持されること,一方,TCI-9およびTCI-10はMKNKとの結合能が低下しているためMKNKの活性が高く維持され,この負のフィードバックループが機能することによりRAS-MAPKシグナル伝達経路が抑制されることも見い出された.
以上の結果から,化学遺伝学的なスクリーニングおよびin silicoモデリング解析から明らかにされたソラフェニブの類縁体におけるキャップの大型化がショウジョウバエの甲状腺髄様がんモデルにおける抗腫瘍効果の増強に直結すると結論された.
5.TCI-10はヒトの甲状腺髄様がんを標準的な治療薬よりも強力に抑制する
甲状腺髄様がんに対するTCI-10の抗腫瘍効果についてさらに検討するため,ヒトの甲状腺髄様がんに由来するTT細胞をヌードマウスの皮下に移植して腫瘍を形成させ,TCI-10,ソラフェニブ,現在の甲状腺髄様がんの標準的な治療薬であるカボザンチニブを投与した.その結果,TCI-10は投与されたマウスの75%に部分寛解や完全寛解をもたらした.この抗腫瘍効果は,寛解をまったくもたらさなかったソラフェニブやカボザンチニブよりもはるかに強力なものであった.また,TCI-10はマウスにおける副作用を発現しない最大投与量が一般の薬物よりも高く,30日間の投与によりマウスの体重や行動に影響をおよぼさなかった.これらの結果から,TCI-10は抗甲状腺髄様がんに対する抗腫瘍効果およびin vivoにおける好ましい動態を兼ね備えたリード化合物であることが示された.
おわりに
この研究において,筆者らは,ショウジョウバエにおける化学遺伝学的なスクリーニングとin silicoモデリング解析とを融合し,認可薬から論理的に新規のリード化合物を創出する手法を開発した.おもに,ソラフェニブの類縁体の合成,anti-targetの同定,anti-targetと結合しない化学構造の導出,の3段階で甲状腺髄様がんの動物モデルにおいて高い抗腫瘍効果を示すリード化合物を創出した(図1).大きな特色として,ショウジョウバエを創薬の研究に活用することにより,遺伝学的な網羅的な解析および個体における迅速な薬効の評価が可能となった点,そして,異分野融合により効率的な基盤が確立された点をあげたい.
また,化学的な観点から,F原子による修飾が抗腫瘍効果の増強に有効であった点は興味深い.近年,F原子による既存薬の修飾は大きな注目をあつめているためである.実際,複数のF原子をもつ認可薬が増加してきており,7つのF原子をもつエンザルタミドやアプレピタントなどはその例である10).F原子の物性がどのように薬効に影響するかは完全には解明されていないものの,この研究において示されたC-F鎖による修飾は今後も化学構造の改変の有効な手段になる可能性がある.
これまで,がんの創薬の分野においては,単一の標的の阻害をめざす分子標的薬が大きな注目をあつめてきた.この研究において示された創薬の基盤はこれと相補的なものであり,低費用で効果的に新規のリード化合物を創出することが可能になると期待される.さらに,がんの発生を促進するキナーゼシグナル伝達ネットワークに対し多重薬理学的な化合物が有効であることも示された.この手法は,神経疾患や心疾患など創薬の困難な疾患にも適用できる可能性があり,創薬の分野を大きく変革する可能性をひめている.今後も,この手法の発展と応用をつうじ,がんをはじめとする疾患に苦しむ人たちの救済に貢献したい.
文 献
- Hay, M., Thomas, D. W., Craighead, J. L. et al.: Clinical development success rates for investigational drugs. Nat. Biotechnol., 32, 40-51 (2014)[PubMed]
- Knight, Z. A., Lin, H. & Shokat, K. M.: Targeting the cancer kinome through polypharmacology. Nat. Rev. Cancer, 10, 130-137 (2010)[PubMed]
- Davis, M. I., Hunt, J. P., Herrgard, S. et al.: Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol., 29, 1046-1051 (2011)[PubMed]
- Dar, A. C., Das, T. K., Shokat, K. M. et al.: Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature, 486, 80-84 (2012)[PubMed]
- Sonoshita, M. & Cagan, R. L.: Modeling human cancers in Drosophila. Curr. Top. Dev. Biol., 121, 287-309 (2017)[PubMed]
- Carlomagno, F., Anaganti, S., Guida, T. et al.: BAY 43-9006 inhibition of oncogenic RET mutants. J. Natl. Cancer Inst., 98, 326-334 (2006)[PubMed]
- Lam, E. T., Ringel, M. D., Kloos, R. T. et al.: Phase II clinical trial of sorafenib in metastatic medullary thyroid cancer. J. Clin. Oncol., 28, 2323-2330 (2010)[PubMed]
- Ung, P. M. & Schlessinger, A.: DFGmodel: predicting protein kinase structures in inactive states for structure-based discovery of type-II inhibitors. ACS Chem. Biol., 10, 269-278 (2015)[PubMed]
- Huang, A. M. & Rubin, G. M.: A misexpression screen identifies genes that can modulate RAS1 pathway signaling in Drosophila melanogaster. Genetics, 156, 1219-1230 (2000)[PubMed]
- Muller, K., Faeh, C. & Diederich, F.: Fluorine in pharmaceuticals: looking beyond intuition. Science, 317, 1881-1886 (2007)[PubMed]
活用したデータベースにかかわるキーワードと統合TVへのリンク
著者プロフィール
略歴:2004年 京都大学大学院医学研究科 修了,同 助教,同 講師,同 准教授を経て,2013年より米国Icahn School of Medicine at Mount Sinai研究員.
研究テーマ:がんの発生の機序の解明およびがんの治療薬の創出.
Ross L. Cagan
米国Icahn School of Medicine at Mount SinaiにてProfessor.
研究室URL:http://drb.mssm.edu/labs/cagan.html
Arvin C. Dar
米国Icahn School of Medicine at Mount SinaiにてAssistant Professor.
研究室URL:http://labs.icahn.mssm.edu/darlab/
© 2018 園下将大・Ross L. Cagan・Arvin C. Dar Licensed under CC 表示 2.1 日本
