Hajdu-Cheney症候群型のNOTCH2変異体はSCFFBW7ユビキチンリガーゼ複合体に依存的なタンパク質分解からの逸脱により骨粗鬆症の病態をひき起こす
福島秀文1・Wenyi Wei 2・犬塚博之1
(1東北大学大学院歯学研究科 先端再生医学研究センター,2米国Harvard Medical School,Beth Israel Deaconess Medical Center)
email:福島秀文,犬塚博之
DOI: 10.7875/first.author.2017.137
NOTCH2 Hajdu-Cheney mutations escape SCFFBW7-dependent proteolysis to promote osteoporosis.
Hidefumi Fukushima, Kouhei Shimizu, Asami Watahiki, Seira Hoshikawa, Tomoki Kosho, Daiju Oba, Seiji Sakano, Makiko Arakaki, Aya Yamada, Katsuyuki Nagashima, Koji Okabe, Satoshi Fukumoto, Eijiro Jimi, Anna Bigas, Keiichi I. Nakayama, Keiko Nakayama, Yoko Aoki, Wenyi Wei, Hiroyuki Inuzuka
Molecular Cell, 68, 645-658.e5 (2017)
Hajdu-Cheney症候群は末節骨の骨吸収,進行性の骨粗鬆症,頭蓋骨の変形をおもな病態とする稀少な先天性の骨疾患である.その原因としてNOTCH2遺伝子の変異が報告されているが,この変異がどのようにHajdu-Cheney症候群の発症につながるのか,病態の詳細な分子機序は明らかにされていない.この研究において,筆者らは,Hajdu-Cheney症候群型のNOTCH2変異体はSCFFBW7ユビキチンリガーゼ複合体によるユビキチン化を介したプロテアソームに依存的なタンパク質分解から逸脱し,安定化されたNOTCH2変異体が破骨細胞の分化を亢進させ過剰な骨吸収をひき起こすことを明らかにした.このことから,破骨細胞の分化におけるNOTCH2の分解の制御機構の異常がHajdu-Cheney症候群における骨粗鬆症の病態の一因になること,そして,変異により恒常的に活性化したNOTCH2シグナル伝達経路がこれら骨疾患に対する治療の有効な標的となりうることが示唆された.
骨の健康を維持するため骨の内側では形成と破壊とが恒常的にくり返される骨のリモデリングが起こっている.この骨リモデリングを構成するのは,骨の形成を促進する骨芽細胞および骨を破壊し吸収する破骨細胞である.これら骨芽細胞と破骨細胞とのあいだの骨形成の活性と骨吸収の活性の正常なバランスが骨の恒常性の維持において重要である.進行性の骨融解を主要な病態とするHajdu-Cheney症候群においては,破骨細胞による骨吸収の活性の異常な亢進がその病態の原因であることが示唆されていた.そして,Hajdu-Cheney症候群の患者を対象とした全エクソーム解析からNOTCH2遺伝子の変異が報告され原因遺伝子として同定された1,2).
NOTCH2は細胞のあいだのシグナル伝達を仲介する1回膜貫通型タンパク質であり,細胞外ドメインが隣接する細胞の細胞膜に存在するリガンドDelta/Jaggedと結合することにより細胞内ドメインがプロセシングをうけて核へと移行し,下流の標的となる遺伝子の発現を制御する3).4種類が報告されているNOTCHのなかでも,NOTCH2は血管の形成および腎臓や肝臓の発生に重要であることが報告されているが,筆者らの研究グループは,とくに骨代謝における役割に焦点をあてており,以前に,NOTCH2シグナル伝達経路が破骨細胞の分化に重要であることを報告している4).この研究においては,NOTCH2遺伝子の変異によりひき起こされるHajdu-Cheney症候群の病態について分子機序の詳細を明らかにすることを目的とした.
Hajdu-Cheney症候群の患者におけるNOTCH2遺伝子の変異はC末端側の最終エキソンに位置するPEST配列あるいはその上流に集中している1,2).それらはミスセンス変異およびフレームシフト変異を主とすることから,Hajdu-Cheney症候群型のNOTCH2変異体はPEST配列を欠失していると想定されている.PEST配列はタンパク質分解をひき起こすのに必要な情報をもつことから,Hajdu-Cheney症候群型のNOTCH2変異体において欠失している領域の塩基配列を精査したところ,SCFFBW7ユビキチンリガーゼ複合体の基質認識タンパク質であるFBW7(図1)に対するコンセンサスな認識配列が存在することを見い出した.FBW7は細胞の増殖あるいは生存に重要なタンパク質と選択的に結合することにより,それらのユビキチン化およびタンパク質分解を制御する主要ながん抑制遺伝子産物としての機能が知られている5,6).
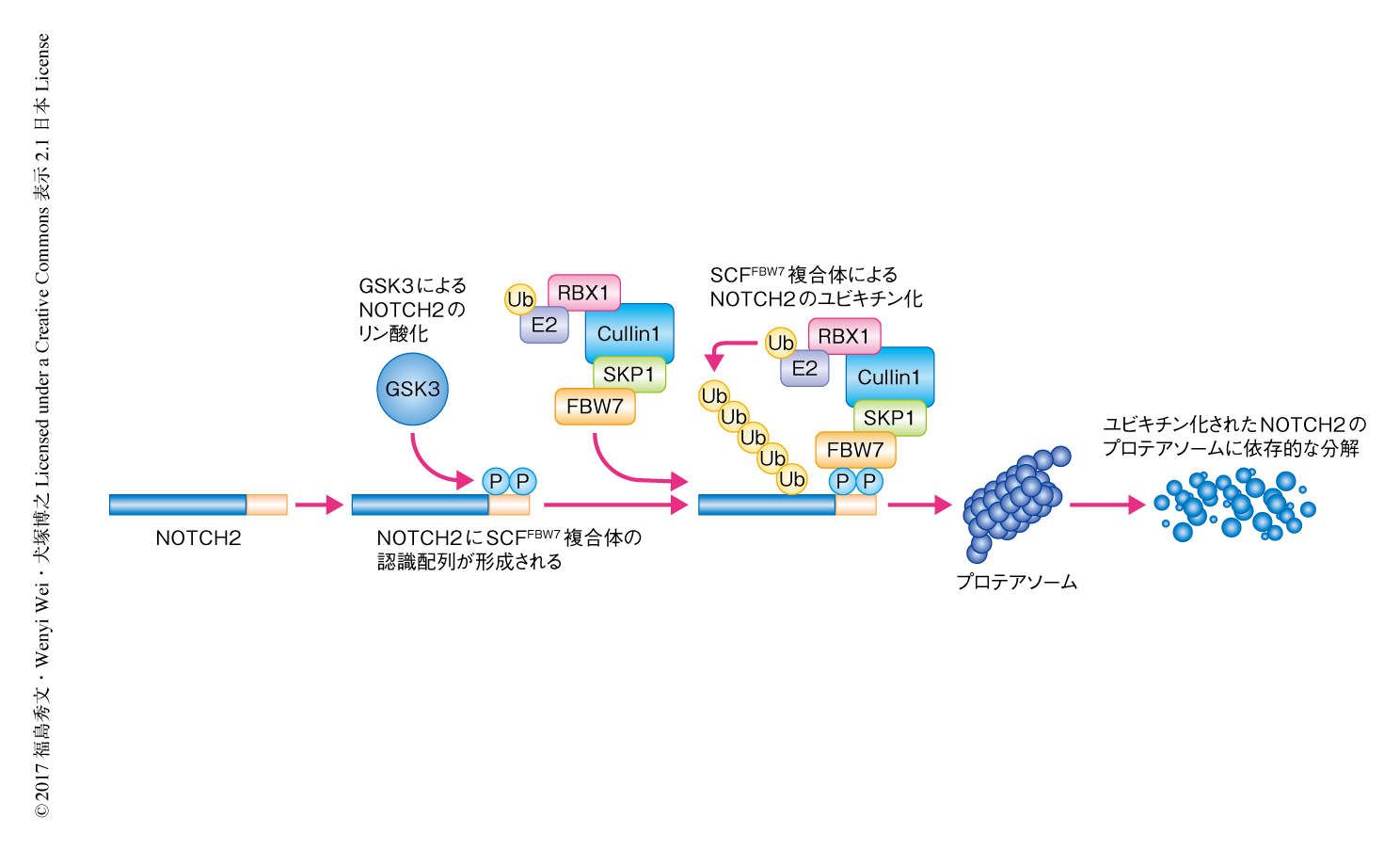
Hajdu-Cheney症候群型のNOTCH2変異体の解析にさきだち,野生型のNOTCH2とFBW7との相互作用について,免疫沈降法,また,ユビキチン化およびタンパク質分解の解析など生化学的な手法により検討したところ,NOTCH2はGSK3によるリン酸化を介したFBW7との結合によりユビキチン化され,プロテアソームに依存的な分解をうけることが確認された(図1).FBW7の認識配列を欠失しているHajdu-Cheney症候群型のNOTCH2変異体を用いて同様に解析したところ,FBW7との結合能は消失しており,FBW7を介したユビキチン化およびプロテアソームに依存的な分解をうけず安定化した.これらのことから,Hajdu-Cheney症候群型のNOTCH2変異体はFBW7を介したタンパク質分解から逸脱することにより安定化して細胞に蓄積し,NOTCH2の下流のシグナル伝達経路を活性化することが示唆された.
筆者らによる,NOTCH2は破骨細胞の形成を促進する機能をもつという報告4) と一致して,別の研究グループから,Hajdu-Cheney症候群型のNOTCH2変異のノックインマウスは骨吸収の活性化にともなう骨量の減少を示すことが報告された7).これらの背景から,Hajdu-Cheney症候群型のNOTCH2変異体の骨髄前駆細胞への強制発現が破骨細胞の形成にどのように影響をおよぼすか検討した.マウスの骨髄前駆細胞においてNotch2をノックダウンしたところ破骨細胞の形成は抑制された.Notch2をノックダウンした細胞に野生型のNotch2を再導入したところ破骨細胞の形成は回復したが,野生型のNotch2を再導入した細胞との比較において,Hajdu-Cheney症候群型のNotch2変異体を再導入した細胞においては破骨細胞の形成のさらなる亢進が認められた.
臨床学的な観点から,Hajdu-Cheney症候群型のNOTCH2変異体が実際に患者の骨髄前駆細胞において活性化されているかどうか確認した.Hajdu-Cheney症候群型の患者より採取した末梢血細胞において,破骨細胞が形成される際のNOTCH2 mRNAの発現量の比較においては健常者と患者とのあいだに差はみられなかったが,患者においては破骨細胞の形成にともなうNOTCH2の蓄積量の増加が認められた.さらに,NOTCH2の安定性について解析したところ,Hajdu-Cheney症候群の患者から採取した細胞においてNOTCH2の半減期に明らかな延長が認められた.以上の結果から,FBW7を介したタンパク質分解から逸脱して安定化したHajdu-Cheney症候群型のNOTCH2変異体は,細胞における蓄積を介して下流のシグナル伝達経路を活性化させ,破骨細胞の分化を亢進させることが示された.
Hajdu-Cheney症候群において観察される病態がFBW7を介したNOTCH2の分解の異常に起因することが示唆されたことから,破骨細胞に特異的なFbw7のコンディショナルノックアウトマウスを作製することによりFbw7-Notch2シグナル伝達経路が生体における骨代謝に重要であることの証明を試みた.破骨細胞に特異的なFbw7ノックアウトマウスにおいては,骨量の減少,破骨細胞の数の増加,骨吸収の活性化が認められた一方,骨芽細胞の分化のパラメーターに変化はみられなかった.さらに,X線像において明らかな骨吸収が確認され,骨端部における骨融解の進行および手指骨の骨破壊が観察された.また,大腿骨と脛骨を用いた3点荷重試験において明らかな骨強度の低下が確認された.以上の結果から,破骨細胞に特異的なFbw7の欠失により破骨細胞の増加およびその活性化がひき起こされることが示唆された.
破骨細胞に特異的なFbw7ノックアウトマウスにおいてみられた骨吸収の活性化は破骨細胞の分化の亢進に起因することを確認するため,破骨細胞に特異的なFbw7ノックアウトマウスに由来する骨髄前駆細胞において破骨細胞を分化させた.その結果,野生型のマウスと比較して破骨細胞の形成に有意な亢進が認められ,その際には,Notch2が蓄積して下流の標的となる遺伝子の発現が誘導され,破骨細胞の分化の指標であるc-FosやNFATc1の増加,Fbw7の既報の基質であるNF-κB2の活性化が観察された.また,破骨細胞に特異的なFbw7ノックアウトマウスにおける破骨細胞の分化の亢進がNotch2の活性化によるものであることを証明するため,破骨細胞に特異的なFbw7ノックアウトマウスに由来する骨髄前駆細胞においてNotch2あるいはNotch1をそれぞれノックダウンして破骨細胞を分化させたところ,Notch2をノックダウンした細胞においてのみ破骨細胞の過剰な形成が有意に抑制された.以上の結果から,破骨細胞に特異的なFbw7ノックアウトマウスにおいて観察された骨吸収の活性化は,破骨細胞前駆細胞におけるNotch2の安定化とそれにともなう破骨細胞の分化の亢進によることが確認された.さらに,破骨細胞に特異的なFbw7ノックアウトマウスの表現型の多くはHajdu-Cheney症候群の病態と一致した.
NOTCHシグナル伝達経路の効率的な遮断にはNOTCHの細胞内ドメインをプロセシングするγセクレターゼの阻害が有効なアプローチとして用いられている.γセクレターゼの活性を効果的に阻害する低分子化合物DAPTは変形性の骨関節炎に対し治療の効果のあることが疾患モデルマウスの解析により明らかにされ,NOTCHシグナルはDAPTによる骨融解性疾患の治療の標的であることが示された8).そこで,DAPTの投与によるHajdu-Cheney症候群の治療法を開発することを目的として,破骨細胞に特異的なFbw7ノックアウトマウスと薬理学的な手法とを組み合わせた解析系を用いてDAPTの薬効を評価した.解析においては,DAPTのほか,破骨細胞のはたらきを抑制することにより骨吸収の阻害活性を示す臨床治療薬であるゾレドロン酸を用いた.その結果,DAPTはゾレドロン酸と同様に,破骨細胞に特異的なFbw7ノックアウトマウスに由来する骨髄前駆細胞の破骨細胞の形成をin vitroにおいて有意に抑制した.さらに,マウスへ直接に投与した際にも同様に,破骨細胞に特異的なFbw7ノックアウトマウスにおいて骨吸収の抑制および骨吸収のパラメーターの改善が認められ,骨量の回復が観察された.以上の薬理学的な解析から,破骨細胞に特異的なFbw7ノックアウトマウスにおいて観察される骨吸収の活性化がDAPTの投与により抑制されることが示され,FBW7-NOTCH2シグナル伝達経路がHajdu-Cheney症候群の治療の有効な標的になる可能性が提示された.
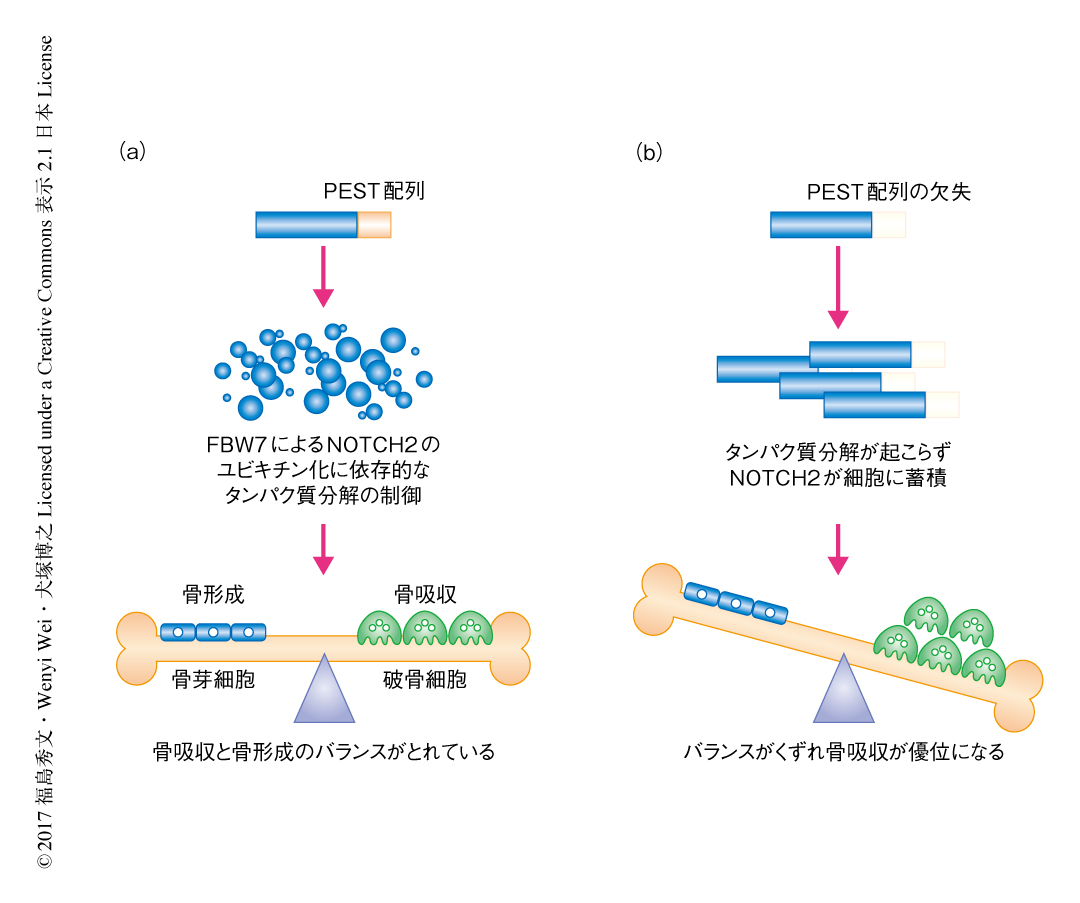
この研究により,NOTCH2遺伝子の変異にともなうHajdu-Cheney症候群の発症について分子機序の一端が明らかにされた(図2).この研究においては,破骨細胞の分化に焦点をあてたが,Hajdu-Cheney症候群における破骨細胞の異常な分化は,破骨細胞そのものの分化活性の上昇のほか,骨芽細胞からの過剰なRANKLの供給によるとの報告もある9).したがって,Hajdu-Cheney症候群におけるNOTCH2遺伝子の変異と,骨芽細胞と破骨細胞とのコミュニケーションに影響をおよぼすほかの臓器との関係も,今後の研究課題になると考えられる.Hajdu-Cheney症候群は稀少性疾患ながら,その分子病態は高齢化社会において高い罹患率を示す骨粗鬆症にも深く関連することが想定される.この研究において得られた知見が,Hajdu-Cheney症候群や骨粗鬆症に対する有効な治療法の開発に貢献することが期待される.
略歴:2005年 福岡歯科大学大学院歯学研究科 修了,同年 福岡歯科大学 助手,2007年 九州歯科大学 助教,2010年 米国Harvard Medical School研究員,2012年 九州歯科大学 助教,同 講師,2013年 福岡歯科大学 准教授を経て,2015年より東北大学大学院歯学研究科 准教授.
研究テーマ:骨代謝および発がんに関連するユビキチンリガーゼの機能.
Wenyi Wei
米国Harvard Medical SchoolにてProfessor.
犬塚 博之(Hiroyuki Inuzuka)
東北大学大学院歯学研究科 准教授.
© 2017 福島秀文・Wenyi Wei・犬塚博之 Licensed under CC 表示 2.1 日本
(1東北大学大学院歯学研究科 先端再生医学研究センター,2米国Harvard Medical School,Beth Israel Deaconess Medical Center)
email:福島秀文,犬塚博之
DOI: 10.7875/first.author.2017.137
NOTCH2 Hajdu-Cheney mutations escape SCFFBW7-dependent proteolysis to promote osteoporosis.
Hidefumi Fukushima, Kouhei Shimizu, Asami Watahiki, Seira Hoshikawa, Tomoki Kosho, Daiju Oba, Seiji Sakano, Makiko Arakaki, Aya Yamada, Katsuyuki Nagashima, Koji Okabe, Satoshi Fukumoto, Eijiro Jimi, Anna Bigas, Keiichi I. Nakayama, Keiko Nakayama, Yoko Aoki, Wenyi Wei, Hiroyuki Inuzuka
Molecular Cell, 68, 645-658.e5 (2017)
要 約
Hajdu-Cheney症候群は末節骨の骨吸収,進行性の骨粗鬆症,頭蓋骨の変形をおもな病態とする稀少な先天性の骨疾患である.その原因としてNOTCH2遺伝子の変異が報告されているが,この変異がどのようにHajdu-Cheney症候群の発症につながるのか,病態の詳細な分子機序は明らかにされていない.この研究において,筆者らは,Hajdu-Cheney症候群型のNOTCH2変異体はSCFFBW7ユビキチンリガーゼ複合体によるユビキチン化を介したプロテアソームに依存的なタンパク質分解から逸脱し,安定化されたNOTCH2変異体が破骨細胞の分化を亢進させ過剰な骨吸収をひき起こすことを明らかにした.このことから,破骨細胞の分化におけるNOTCH2の分解の制御機構の異常がHajdu-Cheney症候群における骨粗鬆症の病態の一因になること,そして,変異により恒常的に活性化したNOTCH2シグナル伝達経路がこれら骨疾患に対する治療の有効な標的となりうることが示唆された.
はじめに
骨の健康を維持するため骨の内側では形成と破壊とが恒常的にくり返される骨のリモデリングが起こっている.この骨リモデリングを構成するのは,骨の形成を促進する骨芽細胞および骨を破壊し吸収する破骨細胞である.これら骨芽細胞と破骨細胞とのあいだの骨形成の活性と骨吸収の活性の正常なバランスが骨の恒常性の維持において重要である.進行性の骨融解を主要な病態とするHajdu-Cheney症候群においては,破骨細胞による骨吸収の活性の異常な亢進がその病態の原因であることが示唆されていた.そして,Hajdu-Cheney症候群の患者を対象とした全エクソーム解析からNOTCH2遺伝子の変異が報告され原因遺伝子として同定された1,2).
NOTCH2は細胞のあいだのシグナル伝達を仲介する1回膜貫通型タンパク質であり,細胞外ドメインが隣接する細胞の細胞膜に存在するリガンドDelta/Jaggedと結合することにより細胞内ドメインがプロセシングをうけて核へと移行し,下流の標的となる遺伝子の発現を制御する3).4種類が報告されているNOTCHのなかでも,NOTCH2は血管の形成および腎臓や肝臓の発生に重要であることが報告されているが,筆者らの研究グループは,とくに骨代謝における役割に焦点をあてており,以前に,NOTCH2シグナル伝達経路が破骨細胞の分化に重要であることを報告している4).この研究においては,NOTCH2遺伝子の変異によりひき起こされるHajdu-Cheney症候群の病態について分子機序の詳細を明らかにすることを目的とした.
1.Hajdu-Cheney症候群型のNOTCH2変異体はプロテアソームに依存的なタンパク質分解から逸脱して安定化することにより破骨細胞の分化を亢進させる
Hajdu-Cheney症候群の患者におけるNOTCH2遺伝子の変異はC末端側の最終エキソンに位置するPEST配列あるいはその上流に集中している1,2).それらはミスセンス変異およびフレームシフト変異を主とすることから,Hajdu-Cheney症候群型のNOTCH2変異体はPEST配列を欠失していると想定されている.PEST配列はタンパク質分解をひき起こすのに必要な情報をもつことから,Hajdu-Cheney症候群型のNOTCH2変異体において欠失している領域の塩基配列を精査したところ,SCFFBW7ユビキチンリガーゼ複合体の基質認識タンパク質であるFBW7(図1)に対するコンセンサスな認識配列が存在することを見い出した.FBW7は細胞の増殖あるいは生存に重要なタンパク質と選択的に結合することにより,それらのユビキチン化およびタンパク質分解を制御する主要ながん抑制遺伝子産物としての機能が知られている5,6).
Hajdu-Cheney症候群型のNOTCH2変異体の解析にさきだち,野生型のNOTCH2とFBW7との相互作用について,免疫沈降法,また,ユビキチン化およびタンパク質分解の解析など生化学的な手法により検討したところ,NOTCH2はGSK3によるリン酸化を介したFBW7との結合によりユビキチン化され,プロテアソームに依存的な分解をうけることが確認された(図1).FBW7の認識配列を欠失しているHajdu-Cheney症候群型のNOTCH2変異体を用いて同様に解析したところ,FBW7との結合能は消失しており,FBW7を介したユビキチン化およびプロテアソームに依存的な分解をうけず安定化した.これらのことから,Hajdu-Cheney症候群型のNOTCH2変異体はFBW7を介したタンパク質分解から逸脱することにより安定化して細胞に蓄積し,NOTCH2の下流のシグナル伝達経路を活性化することが示唆された.
筆者らによる,NOTCH2は破骨細胞の形成を促進する機能をもつという報告4) と一致して,別の研究グループから,Hajdu-Cheney症候群型のNOTCH2変異のノックインマウスは骨吸収の活性化にともなう骨量の減少を示すことが報告された7).これらの背景から,Hajdu-Cheney症候群型のNOTCH2変異体の骨髄前駆細胞への強制発現が破骨細胞の形成にどのように影響をおよぼすか検討した.マウスの骨髄前駆細胞においてNotch2をノックダウンしたところ破骨細胞の形成は抑制された.Notch2をノックダウンした細胞に野生型のNotch2を再導入したところ破骨細胞の形成は回復したが,野生型のNotch2を再導入した細胞との比較において,Hajdu-Cheney症候群型のNotch2変異体を再導入した細胞においては破骨細胞の形成のさらなる亢進が認められた.
臨床学的な観点から,Hajdu-Cheney症候群型のNOTCH2変異体が実際に患者の骨髄前駆細胞において活性化されているかどうか確認した.Hajdu-Cheney症候群型の患者より採取した末梢血細胞において,破骨細胞が形成される際のNOTCH2 mRNAの発現量の比較においては健常者と患者とのあいだに差はみられなかったが,患者においては破骨細胞の形成にともなうNOTCH2の蓄積量の増加が認められた.さらに,NOTCH2の安定性について解析したところ,Hajdu-Cheney症候群の患者から採取した細胞においてNOTCH2の半減期に明らかな延長が認められた.以上の結果から,FBW7を介したタンパク質分解から逸脱して安定化したHajdu-Cheney症候群型のNOTCH2変異体は,細胞における蓄積を介して下流のシグナル伝達経路を活性化させ,破骨細胞の分化を亢進させることが示された.
2.破骨細胞に特異的なFbw7ノックアウトマウスはHajdu-Cheney症候群を模倣した骨粗鬆症様の表現型を呈する
Hajdu-Cheney症候群において観察される病態がFBW7を介したNOTCH2の分解の異常に起因することが示唆されたことから,破骨細胞に特異的なFbw7のコンディショナルノックアウトマウスを作製することによりFbw7-Notch2シグナル伝達経路が生体における骨代謝に重要であることの証明を試みた.破骨細胞に特異的なFbw7ノックアウトマウスにおいては,骨量の減少,破骨細胞の数の増加,骨吸収の活性化が認められた一方,骨芽細胞の分化のパラメーターに変化はみられなかった.さらに,X線像において明らかな骨吸収が確認され,骨端部における骨融解の進行および手指骨の骨破壊が観察された.また,大腿骨と脛骨を用いた3点荷重試験において明らかな骨強度の低下が確認された.以上の結果から,破骨細胞に特異的なFbw7の欠失により破骨細胞の増加およびその活性化がひき起こされることが示唆された.
破骨細胞に特異的なFbw7ノックアウトマウスにおいてみられた骨吸収の活性化は破骨細胞の分化の亢進に起因することを確認するため,破骨細胞に特異的なFbw7ノックアウトマウスに由来する骨髄前駆細胞において破骨細胞を分化させた.その結果,野生型のマウスと比較して破骨細胞の形成に有意な亢進が認められ,その際には,Notch2が蓄積して下流の標的となる遺伝子の発現が誘導され,破骨細胞の分化の指標であるc-FosやNFATc1の増加,Fbw7の既報の基質であるNF-κB2の活性化が観察された.また,破骨細胞に特異的なFbw7ノックアウトマウスにおける破骨細胞の分化の亢進がNotch2の活性化によるものであることを証明するため,破骨細胞に特異的なFbw7ノックアウトマウスに由来する骨髄前駆細胞においてNotch2あるいはNotch1をそれぞれノックダウンして破骨細胞を分化させたところ,Notch2をノックダウンした細胞においてのみ破骨細胞の過剰な形成が有意に抑制された.以上の結果から,破骨細胞に特異的なFbw7ノックアウトマウスにおいて観察された骨吸収の活性化は,破骨細胞前駆細胞におけるNotch2の安定化とそれにともなう破骨細胞の分化の亢進によることが確認された.さらに,破骨細胞に特異的なFbw7ノックアウトマウスの表現型の多くはHajdu-Cheney症候群の病態と一致した.
3.NOTCHの阻害剤の投与により破骨細胞に特異的なFbw7ノックアウトマウスにおける骨量の減少が回復する
NOTCHシグナル伝達経路の効率的な遮断にはNOTCHの細胞内ドメインをプロセシングするγセクレターゼの阻害が有効なアプローチとして用いられている.γセクレターゼの活性を効果的に阻害する低分子化合物DAPTは変形性の骨関節炎に対し治療の効果のあることが疾患モデルマウスの解析により明らかにされ,NOTCHシグナルはDAPTによる骨融解性疾患の治療の標的であることが示された8).そこで,DAPTの投与によるHajdu-Cheney症候群の治療法を開発することを目的として,破骨細胞に特異的なFbw7ノックアウトマウスと薬理学的な手法とを組み合わせた解析系を用いてDAPTの薬効を評価した.解析においては,DAPTのほか,破骨細胞のはたらきを抑制することにより骨吸収の阻害活性を示す臨床治療薬であるゾレドロン酸を用いた.その結果,DAPTはゾレドロン酸と同様に,破骨細胞に特異的なFbw7ノックアウトマウスに由来する骨髄前駆細胞の破骨細胞の形成をin vitroにおいて有意に抑制した.さらに,マウスへ直接に投与した際にも同様に,破骨細胞に特異的なFbw7ノックアウトマウスにおいて骨吸収の抑制および骨吸収のパラメーターの改善が認められ,骨量の回復が観察された.以上の薬理学的な解析から,破骨細胞に特異的なFbw7ノックアウトマウスにおいて観察される骨吸収の活性化がDAPTの投与により抑制されることが示され,FBW7-NOTCH2シグナル伝達経路がHajdu-Cheney症候群の治療の有効な標的になる可能性が提示された.
おわりに
この研究により,NOTCH2遺伝子の変異にともなうHajdu-Cheney症候群の発症について分子機序の一端が明らかにされた(図2).この研究においては,破骨細胞の分化に焦点をあてたが,Hajdu-Cheney症候群における破骨細胞の異常な分化は,破骨細胞そのものの分化活性の上昇のほか,骨芽細胞からの過剰なRANKLの供給によるとの報告もある9).したがって,Hajdu-Cheney症候群におけるNOTCH2遺伝子の変異と,骨芽細胞と破骨細胞とのコミュニケーションに影響をおよぼすほかの臓器との関係も,今後の研究課題になると考えられる.Hajdu-Cheney症候群は稀少性疾患ながら,その分子病態は高齢化社会において高い罹患率を示す骨粗鬆症にも深く関連することが想定される.この研究において得られた知見が,Hajdu-Cheney症候群や骨粗鬆症に対する有効な治療法の開発に貢献することが期待される.
文 献
- Isidor, B., Lindenbaum, P., Pichon, O. et al.: Truncating mutations in the last exon of NOTCH2 cause a rare skeletal disorder with osteoporosis. Nat. Genet., 43, 306-308 (2011)[PubMed]
- Simpson, M. A., Irving, M. D., Asilmaz, E. et al.: Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat. Genet., 43, 303-305 (2011)[PubMed]
- Kopan, R. & Ilagan, M. X.: The canonical Notch signaling pathway: unfolding the activation mechanism. Cell, 137, 216-233 (2009)[PubMed]
- Fukushima, H., Nakao, A., Okamoto, F. et al.: The association of Notch2 and NF-κB accelerates RANKL-induced osteoclastogenesis. Mol. Cell. Biol., 28, 6402-6412 (2008)[PubMed]
- Davis, R. J., Welcker, M. & Clurman, B. E.: Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell, 26, 455-464 (2014)[PubMed]
- Wang, Z., Liu, P., Inuzuka, H. et al.: Roles of F-box proteins in cancer. Nat. Rev. Cancer, 14, 233-247 (2014)[PubMed]
- Canalis, E., Schilling, L., Yee, S. P. et al.: Hajdu Cheney mouse mutants exhibit osteopenia, increased osteoclastogenesis, and bone resorption. J. Biol. Chem., 291, 1538-1551 (2016)[PubMed]
- Hosaka, Y., Saito, T., Sugita, S. et al.: Notch signaling in chondrocytes modulates endochondral ossification and osteoarthritis development. Proc. Natl. Acad. Sci. USA, 110, 1875-1880 (2013)[PubMed]
- Zanotti, S., Yu, J., Sanjay, A. et al.: Sustained Notch2 signaling in osteoblasts, but not in osteoclasts, is linked to osteopenia in a mouse model of Hajdu-Cheney syndrome. J. Biol. Chem., 292, 12232-12244 (2017)[PubMed]
活用したデータベースにかかわるキーワードと統合TVへのリンク
著者プロフィール
略歴:2005年 福岡歯科大学大学院歯学研究科 修了,同年 福岡歯科大学 助手,2007年 九州歯科大学 助教,2010年 米国Harvard Medical School研究員,2012年 九州歯科大学 助教,同 講師,2013年 福岡歯科大学 准教授を経て,2015年より東北大学大学院歯学研究科 准教授.
研究テーマ:骨代謝および発がんに関連するユビキチンリガーゼの機能.
Wenyi Wei
米国Harvard Medical SchoolにてProfessor.
犬塚 博之(Hiroyuki Inuzuka)
東北大学大学院歯学研究科 准教授.
© 2017 福島秀文・Wenyi Wei・犬塚博之 Licensed under CC 表示 2.1 日本
