膵β細胞のPI3キナーゼは複数の分子機構によりインスリンの分泌を制御する
金子和真・植木浩二郎
(東京大学大学院医学系研究科 代謝・栄養病態学)
email:植木浩二郎
DOI: 10.7875/first.author.2010.068
Class IA phosphatidylinositol 3-kinase in pancreatic β cells controls insulin secretion by multiple mechanisms.
Kazuma Kaneko, Kohjiro Ueki, Noriko Takahashi, Shinji Hashimoto, Masayuki Okamoto, Motoharu Awazawa, Yukiko Okazaki, Mitsuru Ohsugi, Kazunori Inabe, Toshihiro Umehara, Masashi Yoshida, Masafumi Kakei, Tadahiro Kitamura, Ji Luo, Rohit N. Kulkarni, C. Ronald Kahn, Haruo Kasai, Lewis C. Cantley, Takashi Kadowaki
Cell Metabolism, 12, 619-632 (2010)
2型糖尿病の病態は末梢臓器のインスリン抵抗性と膵β細胞のインスリン分泌不全によって特徴づけられる.とくに,インスリンの分泌に大きく影響をあたえるβ細胞の量や機能の制御には,膵β細胞自体におけるインスリンシグナルが強く関与していることがこれまでに示されてきた.最近,筆者らは,糖尿病モデルマウスの膵β細胞で実際にインスリンシグナルが障害され2型糖尿病の病態形成に関与していること,また,インスリンシグナルを担う重要なタンパク質であるクラスIAホスファチジルイノシトール3-キナーゼ(PI3キナーゼ)は膵β細胞の量を制御するだけでなく,SNAREタンパク質の発現制御を介したインスリンのエキソサイトーシスやCa2+流入におけるβ細胞どうしの同期を介して膵β細胞の機能をも制御していることを明らかにした.これらの結果から,2型糖尿病においてはインスリンの分泌が低下することが膵β細胞自体のインスリンシグナルを低下させ,このことがますますインスリンの分泌を減弱させる,という悪循環にあるために病態の増悪をきたしていると考えられ,PI3キナーゼを標的とした治療がこの悪循環を断ち切る可能性のあることが示唆された.
2型糖尿病は,肝臓や骨格筋をはじめとする末梢臓器でのインスリン作用の低下(インスリン抵抗性)と,膵β細胞からのインスリン分泌の不全を2つの大きな特徴とする.筆者らをはじめいくつかのグループから,膵β細胞からのインスリン分泌の不全は膵β細胞自体のインスリンシグナルの障害によってもひき起こされることが報告されている.実際,膵β細胞に特異的なインスリン受容体のノックアウトマウスはグルコース応答性インスリン分泌障害および膵β細胞量の減少を呈し1),膵β細胞特異的インスリン受容体およびインスリン様成長因子1(insulin-like growth factor-1:IGF1)受容体のダブルノックアウトマウスは顕著なβ細胞量の減少およびインスリン分泌の低下のため重篤な糖尿病を呈し死亡する2).また,インスリン受容体およびIGF1受容体の主要な下流シグナル伝達タンパク質であるIRS2(IRS:insulin receptor substrate)が膵β細胞の量を制御していること3),PDK1およびAktが膵β細胞の成長や機能に重要であることも報告されている4).これらより,クラスIAホスファチジルイノシトール3-キナーゼ(PI3キナーゼ)はインスリン受容体,IGF1受容体,IRS2の下流でPDK1およびAktを介して膵β細胞の量や機能を制御していることが想定されたが,その詳細な分子機構については明らかではなかった(図1).
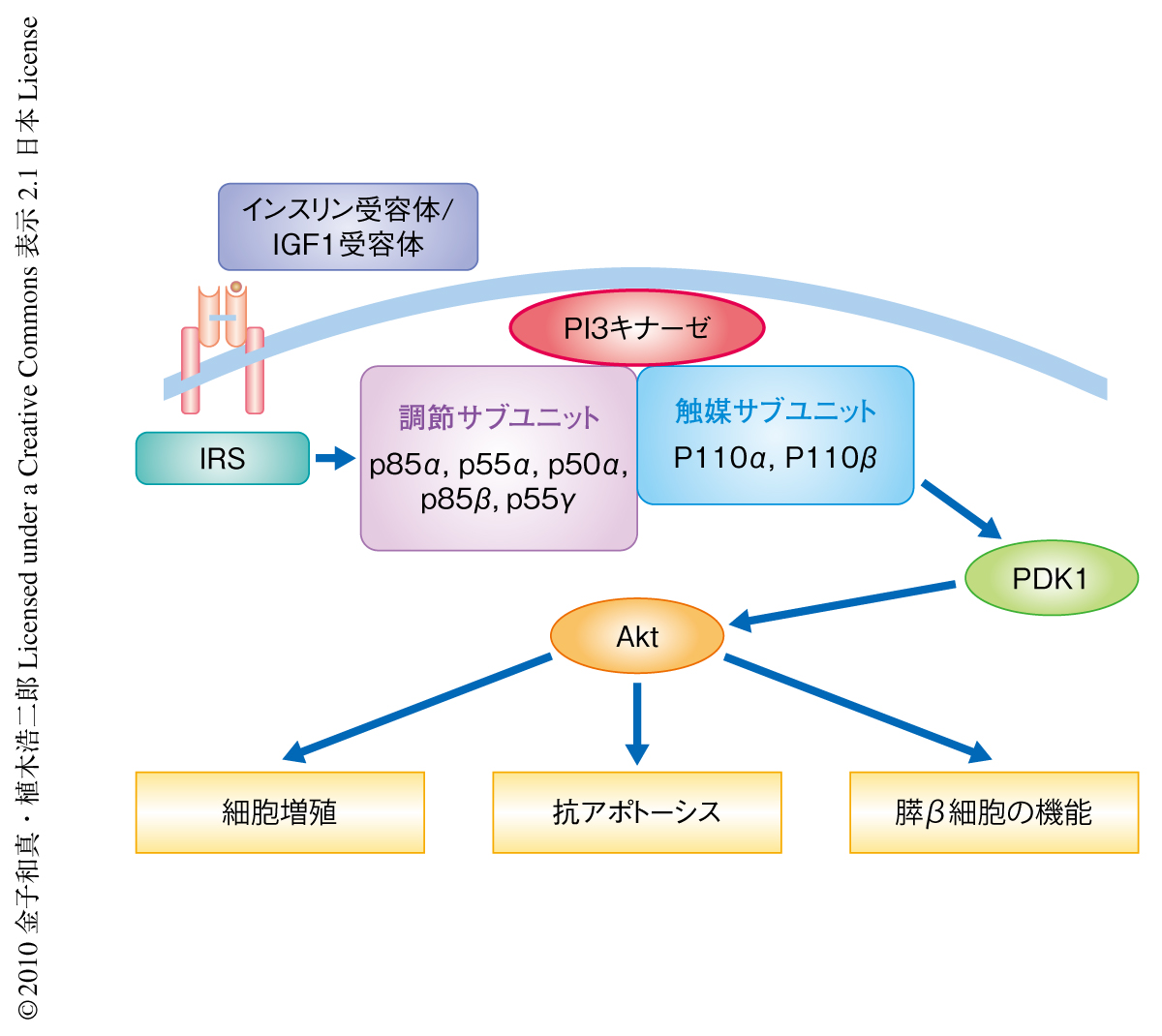
PI3キナーゼは調節サブユニットと触媒サブユニットのヘテロ2量体にて構成され,代謝,増殖作用,抗アポトーシス作用などのシグナルを伝達している.調節サブユニットは3つの異なる遺伝子(pik3r1,pik3r2,pik3r3)によってコードされ,pik3r1遺伝子によって規定されるp85αおよびそのスプライシングバリアントであるp55α,p50α,また,pik3r2遺伝子によって規定されるp85βなどによって構成されている.これらの調節サブユニットのうち,p85αが70%以上,p85βが約20%をしめることが明らかにされており,さまざまな組織におけるPI3キナーゼシグナルの役割を検討するため,肝臓,骨格筋,心臓などをはじめとする臓器で組織特異的なpik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスが作製され報告されてきた5,6).
この研究では,膵β細胞におけるPI3キナーゼの役割を膵β細胞に特異的なPI3キナーゼノックアウトマウスを用いて検討し,また同時に,肥満・糖尿病モデルマウスであるdb/dbマウスを用いて膵β細胞におけるPI3キナーゼの糖尿病の病態における役割について検討を行った.
ヒトではインスリン抵抗性の存在する耐糖能障害の時期にインスリン分泌は一過性に上昇しているが,糖尿病を発症するまえからその分泌は徐々に低下し,発症以降も低下しつづけることが報告されている7).そこで,ヒトの肥満2型糖尿病ときわめてよく似た表現型を呈するdb/dbマウスを用い,膵島におけるインスリンシグナルについて検討を行った.糖尿病の発症まえにはPI3キナーゼを含むインスリンシグナル伝達タンパク質の膵島での発現は上昇しているが,その発現が減少に転じるとインスリン分泌の低下と血糖の上昇とが生じ,糖尿病の発症以降もさらなるインスリンシグナル伝達タンパク質の発現低下とインスリン分泌の低下,高血糖の悪化が認められた.この結果から,PI3キナーゼがインスリン受容体(IGF1受容体)あるいはIRS2の下流でインスリン分泌の制御を介して糖尿病の発症とその悪化に重要な役割を担っている可能性が示唆された.
膵β細胞でのPI3キナーゼの役割を詳細に検討するため,Cre-LoxP系を用いて,膵β細胞に特異的なpik3r1遺伝子ノックアウトマウス,および,全身性pik3r2遺伝子ノックアウトマウスと膵β細胞特異的pik3r1遺伝子ノックアウトマウスとの交配を重ねて膵β細胞に特異的なpik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスを作製した.これらの2つのノックアウトマウスは実際に膵島のウェスタンブロットあるいは免疫組織染色においてもPI3キナーゼのタンパク質レベルでの発現および活性の低下を認め,さらには,PI3キナーゼシグナルが遮断されているためPI3キナーゼの下流でシグナルを伝える転写因子FoxO1の核内に残存しているようすが観察された.以上の結果から,これらのノックアウトマウスでは実際に膵β細胞でPI3キナーゼシグナルの障害されていることが確認された.
膵β細胞特異的pik3r1遺伝子ノックアウトマウスおよび膵β細胞特異的pik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスは正常に発育し,インスリン感受性,随時血糖値,随時インスリン値はその対照と有意差を認めなかった.しかしながら,糖負荷試験を行うとpik3r1遺伝子ノックアウトマウスはグルコース応答性インスリン分泌の低下をともなう耐糖能異常を呈し,pik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスはさらなるインスリン分泌の低下,耐糖能の悪化を呈した.単離した膵島を用いてインスリン分泌についてさらに検討をくわえたところ,高グルコース刺激およびKCl刺激にてこれら2つのノックアウトマウスから単離した膵島からのインスリン分泌は有意に低下しており,膵β細胞においてPI3キナーゼシグナルが障害されるとインスリン分泌の障害されることが示唆された.また,膵β細胞に特異的なCre発現マウスでは視床下部でもCreリコンビナーゼの発現を認めることが報告されている.この研究で用いたノックアウトマウスでもやはり視床下部でのPI3キナーゼの発現は低下していた.視床下部のインスリンシグナルは摂食を制御していることが知られているためpik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスの摂餌量について検討を行ったが,対照と比べて有意な差を認めなかった.
このインスリン分泌の低下は膵β細胞の量の減少あるいは機能の障害により惹起されうる.そこで,まず膵β細胞の量について検討を行った.膵β細胞におけるインスリンシグナルは抗アポトーシス作用などを介して膵β細胞の量を制御していることがすでに報告されている.免疫組織染色における検討では膵β細胞の形態には明らかな異常を認めず,膵β細胞の量(8週齢)には有意な差を認めなかった.しかしながら,TUNEL染色にてアポトーシスについて検討したところ,膵β細胞特異的pik3r1遺伝子ノックアウトマウス,膵β細胞特異的pik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスでは膵島でのTUNEL陽性β細胞が有意に増加していた.アポトーシスが亢進しているにもかかわらず膵β細胞の量には変化を認めなかったため,なんらかの代償機構が存在するのではないかと考えBrdU染色にて細胞増殖について検討を行ったところ,これら2つのノックアウトマウスともに細胞増殖能の亢進が認められた.インスリンシグナルはインスリン受容体の下流でPI3キナーゼ/Aktシグナル経路とRas/Erkシグナル経路の2つのシグナル経路によりその作用を伝える8).PI3キナーゼ/Aktシグナル経路は糖代謝や抗アポトーシス作用などインスリン作用の多くを制御し,一方,Ras/Erkシグナル経路はPI3キナーゼ/Aktシグナル経路と協調しつつ細胞増殖を制御している.そこで,Ras/Erkシグナル経路について検討したところ,2つのノックアウトマウスから単離した膵島ではErkのリン酸化レベルが増加しておりRas/Erkシグナル経路が活性化されていることを確認した.以上より,インスリン受容体(IGF1受容体)あるいはIRSの下流に存在する2つのシグナル経路のうち,PI3キナーゼ/Aktシグナル経路が遮断されるとその負のフィードバックがはずれるためRas/Erkシグナル経路が活性化されて細胞増殖能が亢進し,アポトーシスが亢進しているにもかかわらず膵β細胞の量が維持されるものと考えられた.
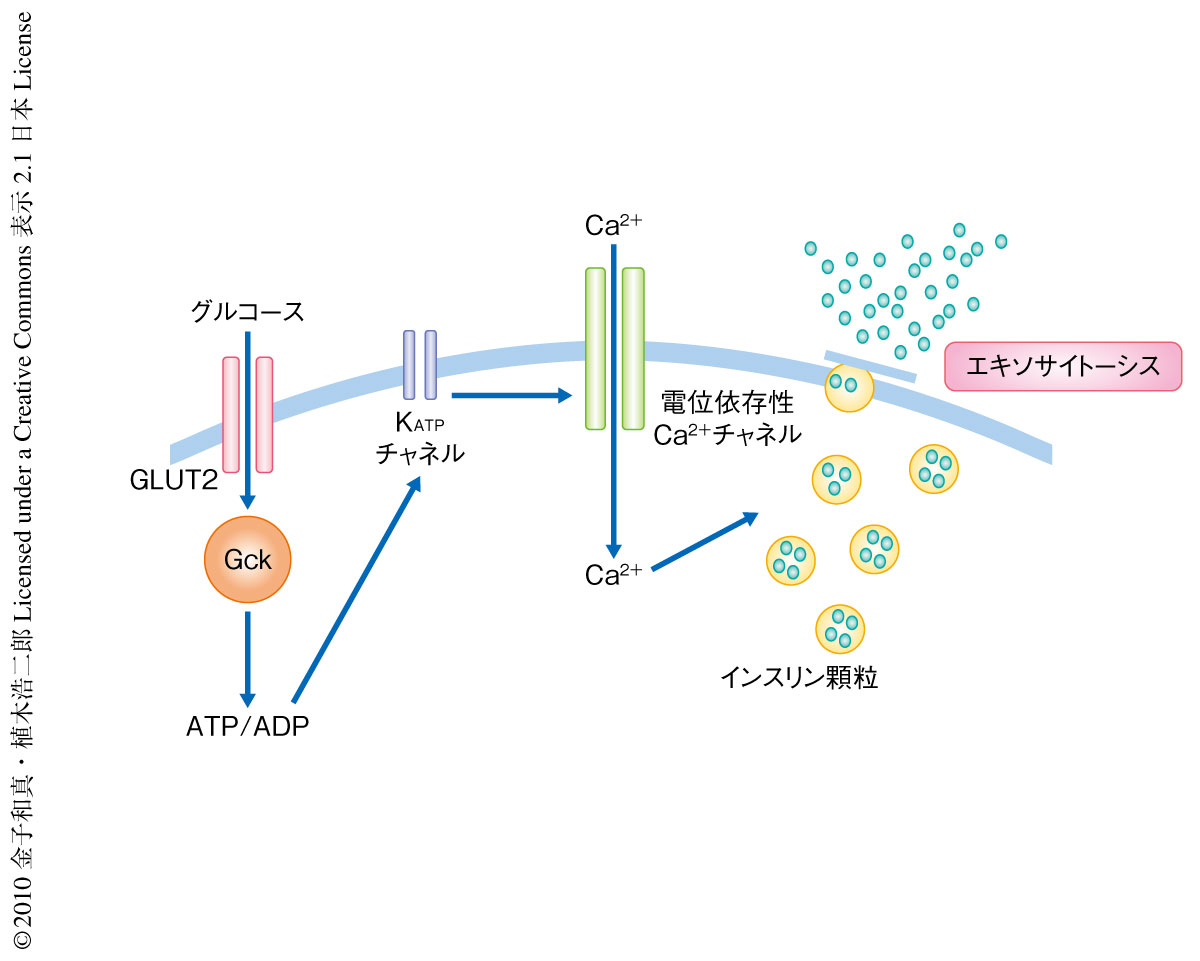
in vivoで認められたグルコース応答性インスリン分泌の障害についてより詳細な検討を行うのに2光子励起法を用いた.2光子励起法ではインスリン顆粒のエキソサイトーシスあるいはCa2+の流入を膵島の状態で観察することができる9).観察の結果,膵β細胞特異的pik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスの膵島ではグルコース刺激によるインスリン顆粒のエキソサイトーシスの回数および膵β細胞へのCa2+の流入量が有意に減少していた.この結果より,糖の取り込みからCa2+流入までのステップまでに障害があることが示唆されたため,インスリン分泌を制御する各ステップについて検討を行った(図2).グルコース刺激による膵島ATP含量には有意な差はないものの減少傾向を呈し,ミトコンドリア関連遺伝子の発現には低下を認め,糖輸送からATP産生までのステップが障害されている可能性が示唆された.つぎに,KATPチャネルのステップについて電気生理学的な解析を行ったものの,電流密度応答性,グルコース応答性,静止膜電位には差を認めなかった.つづいて,Ca2+流入のステップにおいても検討を行ったが,グルコース刺激によるCa2+流入について膵β細胞どうしの同期が障害されているようすが観察された.さらに,このダブルノックアウトマウスではケージドCa2+刺激によるインスリン顆粒のエキソサイトーシスの回数が有意に減少していた.この結果は,Ca2+流入よりも下流のステップ,つまり,インスリンのエキソサイトーシスに障害の存在することを示唆した.以上の結果から,pik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスでは,少なくとも膵β細胞どうしのCa2+流入の同期,および,エキソサイトーシスが障害されてインスリン分泌の低下にいたっているものと考えられた.
さきに述べたエキソサイトーシス,および,膵β細胞どうしのCa2+流入の同期が実際にどのような分子機構によりひき起こされているかにつき検討を行うため,遺伝子発現について検討を行った.インスリン顆粒のエキソサイトーシスはSNAREタンパク質とよばれるSyntaxin1A,SNAP25,VAMP2,Rab27aなどのタンパク質により制御されていることが報告されているが,これらのmRNAおよびタンパク質の発現レベルは膵β細胞特異的pik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスで有意に減少していた.これらSNAREタンパク質の発現低下がこのダブルノックアウトマウスで観察されたインスリン顆粒の分泌障害の一端を担っているものと考えられた.一方,膵β細胞どうしのCa2+流入の同期にはギャップジャンクションが重要な役割を担っていることが報告されている10).膵β細胞のギャップジャンクションはConnexin36というタンパク質の6量体で構成されているが,このConnexin36の発現もやはりpik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスの膵島ではmRNAのレベルおよびタンパク質のレベルで有意に低下しておりCa2+流入の同期障害のひとつの原因であると考えられた.
PI3キナーゼシグナルがこれらの遺伝子発現を実際に制御しているかどうかを検討するためこれらの遺伝子のプロモーター領域について解析を行ったところ,SNAREタンパク質およびConnexin36のすべてのプロモーター領域には転写因子FoxO1の結合配列が認められた.そこで,恒常活性型FoxO1を正常マウスの膵島に発現させこれらの遺伝子の発現を検討したところ,Connexin36とSNAP25では発現レベルが有意に低下し,この2つの遺伝子の発現はFoxO1により負に制御されていることが示唆された.実際に,この2つの遺伝子のプロモーター領域には種をこえて保存されたFoxO1の結合領域が存在していた.また,恒常活性型Aktをpik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスの膵島で発現させてPI3キナーゼ/Aktシグナル経路を回復させこれらの遺伝子の発現を検討したところ,SNAREタンパク質およびConnexin36の発現は増加し,さらには,実際にグルコース応答性インスリン分泌も回復した.これらの結果から,PI3キナーゼシグナルはエキソサイトーシスおよび膵β細胞どうしのCa2+流入の同期を担う遺伝子の発現を制御しインスリン分泌を保っていることが示唆された.
膵β細胞特異的pik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスで認められた膵β細胞どうしのCa2+流入の同期あるいはエキソサイトーシスの障害が糖尿病の病態に関与しているかどうかを検討するため,再びdb/dbマウスを用いた検討を行った.2光子励起法による観察ではこのダブルノックアウトマウスと同様にdb/dbマウスの膵島でもグルコース応答性インスリン分泌は減少し,Ca2+流入の同期障害も認められた.さらに,Connexin36およびSNAREタンパク質の発現はdb/dbマウスのインスリン分泌の低下にともない減少していた.これらの結果から,実際に糖尿病の病態においても糖尿病が悪化してインスリン分泌の低下するにしたがい,パラクラインあるいはオートクラインにより作用するインスリンが減少して,その結果,膵β細胞のインスリン受容体,IRS2,PI3キナーゼ,Aktとつづくシグナルが減弱し,エキソサイトーシスや膵β細胞どうしのCa2+流入の同期を制御する遺伝子の発現が減少する.これらの変化により,最終的にはさらなるインスリン分泌の低下とそれにつづく糖尿病の増悪という悪循環を形成していることが示唆された.
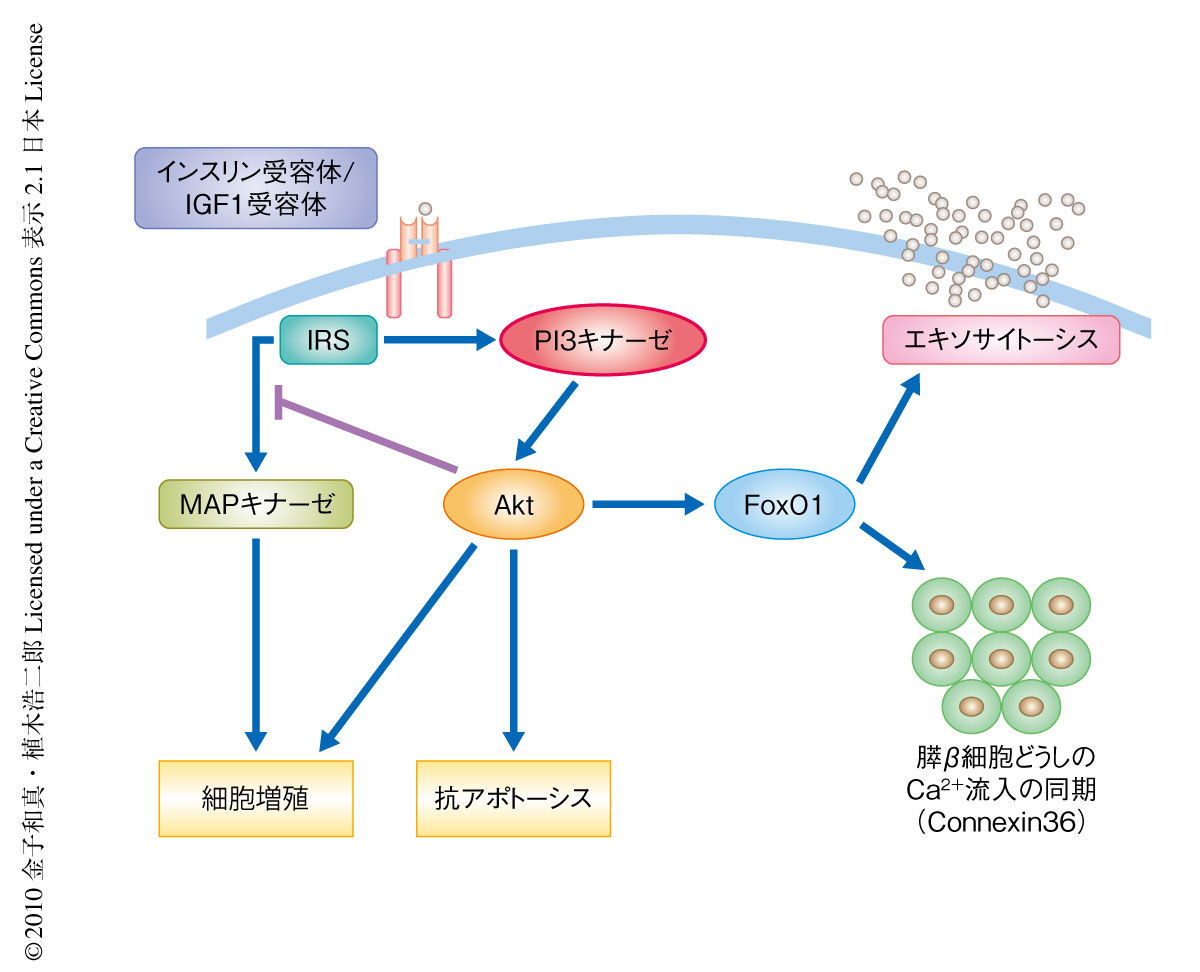
この研究で筆者らは,2型糖尿病の病態においても膵β細胞より分泌されるインスリンはβ細胞自体に作用してその量や機能を制御しており,病態が増悪しインスリン分泌が低下することがβ細胞のインスリンシグナルを低下させ,さらなるβ細胞の量や機能を障害するという悪循環を惹起していること,また,膵β細胞のPI3キナーゼはその量だけではなく,エキソサイトーシスあるいは膵β細胞どうしのCa2+流入の同期を介して膵β細胞の機能を制御していることを明らかにした(図3).この研究で用いた膵β細胞に特異的なPI3キナーゼノックアウトマウスの表現型はインスリン分泌が低下した耐糖能異常,とくに,インスリン初期分泌の障害を呈するなどヒトの2型糖尿病の初期の状態ときわめて類似しており,2型糖尿病の正確な理解に非常に有用であるものと考えられた.この研究により得られた知見から,今後の2型糖尿病への治療戦略として膵β細胞のPI3キナーゼが新規の治療標的タンパク質としてきわめて有望であり,膵β細胞のPI3キナーゼシグナルを上昇させるような薬剤,あるいは,膵β細胞のインスリン抵抗性を改善するような薬剤が新しい可能性を秘めていることが示唆された.
略歴:2010年 東京大学大学院医学系研究科博士課程 修了,同年より東京大学医学部附属病院 特任研究員.
研究テーマ:膵β細胞におけるPI3キナーゼの役割.
関心事・抱負:2型糖尿病の病態を正確に理解し,それにもとづいた糖尿病の治療薬が開発されるよう,研究に邁進したいと思います.
植木 浩二郎(Kohjiro Ueki)
東京大学大学院医学系研究科 准教授.
© 2010 金子和真・植木浩二郎 Licensed under CC 表示 2.1 日本
(東京大学大学院医学系研究科 代謝・栄養病態学)
email:植木浩二郎
DOI: 10.7875/first.author.2010.068
Class IA phosphatidylinositol 3-kinase in pancreatic β cells controls insulin secretion by multiple mechanisms.
Kazuma Kaneko, Kohjiro Ueki, Noriko Takahashi, Shinji Hashimoto, Masayuki Okamoto, Motoharu Awazawa, Yukiko Okazaki, Mitsuru Ohsugi, Kazunori Inabe, Toshihiro Umehara, Masashi Yoshida, Masafumi Kakei, Tadahiro Kitamura, Ji Luo, Rohit N. Kulkarni, C. Ronald Kahn, Haruo Kasai, Lewis C. Cantley, Takashi Kadowaki
Cell Metabolism, 12, 619-632 (2010)
要 約
2型糖尿病の病態は末梢臓器のインスリン抵抗性と膵β細胞のインスリン分泌不全によって特徴づけられる.とくに,インスリンの分泌に大きく影響をあたえるβ細胞の量や機能の制御には,膵β細胞自体におけるインスリンシグナルが強く関与していることがこれまでに示されてきた.最近,筆者らは,糖尿病モデルマウスの膵β細胞で実際にインスリンシグナルが障害され2型糖尿病の病態形成に関与していること,また,インスリンシグナルを担う重要なタンパク質であるクラスIAホスファチジルイノシトール3-キナーゼ(PI3キナーゼ)は膵β細胞の量を制御するだけでなく,SNAREタンパク質の発現制御を介したインスリンのエキソサイトーシスやCa2+流入におけるβ細胞どうしの同期を介して膵β細胞の機能をも制御していることを明らかにした.これらの結果から,2型糖尿病においてはインスリンの分泌が低下することが膵β細胞自体のインスリンシグナルを低下させ,このことがますますインスリンの分泌を減弱させる,という悪循環にあるために病態の増悪をきたしていると考えられ,PI3キナーゼを標的とした治療がこの悪循環を断ち切る可能性のあることが示唆された.
はじめに
2型糖尿病は,肝臓や骨格筋をはじめとする末梢臓器でのインスリン作用の低下(インスリン抵抗性)と,膵β細胞からのインスリン分泌の不全を2つの大きな特徴とする.筆者らをはじめいくつかのグループから,膵β細胞からのインスリン分泌の不全は膵β細胞自体のインスリンシグナルの障害によってもひき起こされることが報告されている.実際,膵β細胞に特異的なインスリン受容体のノックアウトマウスはグルコース応答性インスリン分泌障害および膵β細胞量の減少を呈し1),膵β細胞特異的インスリン受容体およびインスリン様成長因子1(insulin-like growth factor-1:IGF1)受容体のダブルノックアウトマウスは顕著なβ細胞量の減少およびインスリン分泌の低下のため重篤な糖尿病を呈し死亡する2).また,インスリン受容体およびIGF1受容体の主要な下流シグナル伝達タンパク質であるIRS2(IRS:insulin receptor substrate)が膵β細胞の量を制御していること3),PDK1およびAktが膵β細胞の成長や機能に重要であることも報告されている4).これらより,クラスIAホスファチジルイノシトール3-キナーゼ(PI3キナーゼ)はインスリン受容体,IGF1受容体,IRS2の下流でPDK1およびAktを介して膵β細胞の量や機能を制御していることが想定されたが,その詳細な分子機構については明らかではなかった(図1).
PI3キナーゼは調節サブユニットと触媒サブユニットのヘテロ2量体にて構成され,代謝,増殖作用,抗アポトーシス作用などのシグナルを伝達している.調節サブユニットは3つの異なる遺伝子(pik3r1,pik3r2,pik3r3)によってコードされ,pik3r1遺伝子によって規定されるp85αおよびそのスプライシングバリアントであるp55α,p50α,また,pik3r2遺伝子によって規定されるp85βなどによって構成されている.これらの調節サブユニットのうち,p85αが70%以上,p85βが約20%をしめることが明らかにされており,さまざまな組織におけるPI3キナーゼシグナルの役割を検討するため,肝臓,骨格筋,心臓などをはじめとする臓器で組織特異的なpik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスが作製され報告されてきた5,6).
この研究では,膵β細胞におけるPI3キナーゼの役割を膵β細胞に特異的なPI3キナーゼノックアウトマウスを用いて検討し,また同時に,肥満・糖尿病モデルマウスであるdb/dbマウスを用いて膵β細胞におけるPI3キナーゼの糖尿病の病態における役割について検討を行った.
1.db/dbマウスの膵島においてインスリンシグナルは障害されている
ヒトではインスリン抵抗性の存在する耐糖能障害の時期にインスリン分泌は一過性に上昇しているが,糖尿病を発症するまえからその分泌は徐々に低下し,発症以降も低下しつづけることが報告されている7).そこで,ヒトの肥満2型糖尿病ときわめてよく似た表現型を呈するdb/dbマウスを用い,膵島におけるインスリンシグナルについて検討を行った.糖尿病の発症まえにはPI3キナーゼを含むインスリンシグナル伝達タンパク質の膵島での発現は上昇しているが,その発現が減少に転じるとインスリン分泌の低下と血糖の上昇とが生じ,糖尿病の発症以降もさらなるインスリンシグナル伝達タンパク質の発現低下とインスリン分泌の低下,高血糖の悪化が認められた.この結果から,PI3キナーゼがインスリン受容体(IGF1受容体)あるいはIRS2の下流でインスリン分泌の制御を介して糖尿病の発症とその悪化に重要な役割を担っている可能性が示唆された.
2.PI3キナーゼの調節サブユニットの欠損はPI3キナーゼシグナルを障害する
膵β細胞でのPI3キナーゼの役割を詳細に検討するため,Cre-LoxP系を用いて,膵β細胞に特異的なpik3r1遺伝子ノックアウトマウス,および,全身性pik3r2遺伝子ノックアウトマウスと膵β細胞特異的pik3r1遺伝子ノックアウトマウスとの交配を重ねて膵β細胞に特異的なpik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスを作製した.これらの2つのノックアウトマウスは実際に膵島のウェスタンブロットあるいは免疫組織染色においてもPI3キナーゼのタンパク質レベルでの発現および活性の低下を認め,さらには,PI3キナーゼシグナルが遮断されているためPI3キナーゼの下流でシグナルを伝える転写因子FoxO1の核内に残存しているようすが観察された.以上の結果から,これらのノックアウトマウスでは実際に膵β細胞でPI3キナーゼシグナルの障害されていることが確認された.
3.膵β細胞に特異的なPI3キナーゼノックアウトマウスは耐糖能異常を呈する
膵β細胞特異的pik3r1遺伝子ノックアウトマウスおよび膵β細胞特異的pik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスは正常に発育し,インスリン感受性,随時血糖値,随時インスリン値はその対照と有意差を認めなかった.しかしながら,糖負荷試験を行うとpik3r1遺伝子ノックアウトマウスはグルコース応答性インスリン分泌の低下をともなう耐糖能異常を呈し,pik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスはさらなるインスリン分泌の低下,耐糖能の悪化を呈した.単離した膵島を用いてインスリン分泌についてさらに検討をくわえたところ,高グルコース刺激およびKCl刺激にてこれら2つのノックアウトマウスから単離した膵島からのインスリン分泌は有意に低下しており,膵β細胞においてPI3キナーゼシグナルが障害されるとインスリン分泌の障害されることが示唆された.また,膵β細胞に特異的なCre発現マウスでは視床下部でもCreリコンビナーゼの発現を認めることが報告されている.この研究で用いたノックアウトマウスでもやはり視床下部でのPI3キナーゼの発現は低下していた.視床下部のインスリンシグナルは摂食を制御していることが知られているためpik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスの摂餌量について検討を行ったが,対照と比べて有意な差を認めなかった.
4.PI3キナーゼの膵β細胞の量にあたえる影響
このインスリン分泌の低下は膵β細胞の量の減少あるいは機能の障害により惹起されうる.そこで,まず膵β細胞の量について検討を行った.膵β細胞におけるインスリンシグナルは抗アポトーシス作用などを介して膵β細胞の量を制御していることがすでに報告されている.免疫組織染色における検討では膵β細胞の形態には明らかな異常を認めず,膵β細胞の量(8週齢)には有意な差を認めなかった.しかしながら,TUNEL染色にてアポトーシスについて検討したところ,膵β細胞特異的pik3r1遺伝子ノックアウトマウス,膵β細胞特異的pik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスでは膵島でのTUNEL陽性β細胞が有意に増加していた.アポトーシスが亢進しているにもかかわらず膵β細胞の量には変化を認めなかったため,なんらかの代償機構が存在するのではないかと考えBrdU染色にて細胞増殖について検討を行ったところ,これら2つのノックアウトマウスともに細胞増殖能の亢進が認められた.インスリンシグナルはインスリン受容体の下流でPI3キナーゼ/Aktシグナル経路とRas/Erkシグナル経路の2つのシグナル経路によりその作用を伝える8).PI3キナーゼ/Aktシグナル経路は糖代謝や抗アポトーシス作用などインスリン作用の多くを制御し,一方,Ras/Erkシグナル経路はPI3キナーゼ/Aktシグナル経路と協調しつつ細胞増殖を制御している.そこで,Ras/Erkシグナル経路について検討したところ,2つのノックアウトマウスから単離した膵島ではErkのリン酸化レベルが増加しておりRas/Erkシグナル経路が活性化されていることを確認した.以上より,インスリン受容体(IGF1受容体)あるいはIRSの下流に存在する2つのシグナル経路のうち,PI3キナーゼ/Aktシグナル経路が遮断されるとその負のフィードバックがはずれるためRas/Erkシグナル経路が活性化されて細胞増殖能が亢進し,アポトーシスが亢進しているにもかかわらず膵β細胞の量が維持されるものと考えられた.
5.2光子励起法による膵β細胞に特異的なPI3キナーゼノックアウトマウスのインスリン分泌の検討
in vivoで認められたグルコース応答性インスリン分泌の障害についてより詳細な検討を行うのに2光子励起法を用いた.2光子励起法ではインスリン顆粒のエキソサイトーシスあるいはCa2+の流入を膵島の状態で観察することができる9).観察の結果,膵β細胞特異的pik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスの膵島ではグルコース刺激によるインスリン顆粒のエキソサイトーシスの回数および膵β細胞へのCa2+の流入量が有意に減少していた.この結果より,糖の取り込みからCa2+流入までのステップまでに障害があることが示唆されたため,インスリン分泌を制御する各ステップについて検討を行った(図2).グルコース刺激による膵島ATP含量には有意な差はないものの減少傾向を呈し,ミトコンドリア関連遺伝子の発現には低下を認め,糖輸送からATP産生までのステップが障害されている可能性が示唆された.つぎに,KATPチャネルのステップについて電気生理学的な解析を行ったものの,電流密度応答性,グルコース応答性,静止膜電位には差を認めなかった.つづいて,Ca2+流入のステップにおいても検討を行ったが,グルコース刺激によるCa2+流入について膵β細胞どうしの同期が障害されているようすが観察された.さらに,このダブルノックアウトマウスではケージドCa2+刺激によるインスリン顆粒のエキソサイトーシスの回数が有意に減少していた.この結果は,Ca2+流入よりも下流のステップ,つまり,インスリンのエキソサイトーシスに障害の存在することを示唆した.以上の結果から,pik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスでは,少なくとも膵β細胞どうしのCa2+流入の同期,および,エキソサイトーシスが障害されてインスリン分泌の低下にいたっているものと考えられた.
6.膵β細胞の機能はPI3キナーゼシグナルにより制御されている
さきに述べたエキソサイトーシス,および,膵β細胞どうしのCa2+流入の同期が実際にどのような分子機構によりひき起こされているかにつき検討を行うため,遺伝子発現について検討を行った.インスリン顆粒のエキソサイトーシスはSNAREタンパク質とよばれるSyntaxin1A,SNAP25,VAMP2,Rab27aなどのタンパク質により制御されていることが報告されているが,これらのmRNAおよびタンパク質の発現レベルは膵β細胞特異的pik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスで有意に減少していた.これらSNAREタンパク質の発現低下がこのダブルノックアウトマウスで観察されたインスリン顆粒の分泌障害の一端を担っているものと考えられた.一方,膵β細胞どうしのCa2+流入の同期にはギャップジャンクションが重要な役割を担っていることが報告されている10).膵β細胞のギャップジャンクションはConnexin36というタンパク質の6量体で構成されているが,このConnexin36の発現もやはりpik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスの膵島ではmRNAのレベルおよびタンパク質のレベルで有意に低下しておりCa2+流入の同期障害のひとつの原因であると考えられた.
PI3キナーゼシグナルがこれらの遺伝子発現を実際に制御しているかどうかを検討するためこれらの遺伝子のプロモーター領域について解析を行ったところ,SNAREタンパク質およびConnexin36のすべてのプロモーター領域には転写因子FoxO1の結合配列が認められた.そこで,恒常活性型FoxO1を正常マウスの膵島に発現させこれらの遺伝子の発現を検討したところ,Connexin36とSNAP25では発現レベルが有意に低下し,この2つの遺伝子の発現はFoxO1により負に制御されていることが示唆された.実際に,この2つの遺伝子のプロモーター領域には種をこえて保存されたFoxO1の結合領域が存在していた.また,恒常活性型Aktをpik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスの膵島で発現させてPI3キナーゼ/Aktシグナル経路を回復させこれらの遺伝子の発現を検討したところ,SNAREタンパク質およびConnexin36の発現は増加し,さらには,実際にグルコース応答性インスリン分泌も回復した.これらの結果から,PI3キナーゼシグナルはエキソサイトーシスおよび膵β細胞どうしのCa2+流入の同期を担う遺伝子の発現を制御しインスリン分泌を保っていることが示唆された.
7.db/dbマウスでも膵β細胞どうしのCa2+流入の同期およびエキソサイトーシスが障害されている
膵β細胞特異的pik3r1遺伝子pik3r2遺伝子ダブルノックアウトマウスで認められた膵β細胞どうしのCa2+流入の同期あるいはエキソサイトーシスの障害が糖尿病の病態に関与しているかどうかを検討するため,再びdb/dbマウスを用いた検討を行った.2光子励起法による観察ではこのダブルノックアウトマウスと同様にdb/dbマウスの膵島でもグルコース応答性インスリン分泌は減少し,Ca2+流入の同期障害も認められた.さらに,Connexin36およびSNAREタンパク質の発現はdb/dbマウスのインスリン分泌の低下にともない減少していた.これらの結果から,実際に糖尿病の病態においても糖尿病が悪化してインスリン分泌の低下するにしたがい,パラクラインあるいはオートクラインにより作用するインスリンが減少して,その結果,膵β細胞のインスリン受容体,IRS2,PI3キナーゼ,Aktとつづくシグナルが減弱し,エキソサイトーシスや膵β細胞どうしのCa2+流入の同期を制御する遺伝子の発現が減少する.これらの変化により,最終的にはさらなるインスリン分泌の低下とそれにつづく糖尿病の増悪という悪循環を形成していることが示唆された.
おわりに
この研究で筆者らは,2型糖尿病の病態においても膵β細胞より分泌されるインスリンはβ細胞自体に作用してその量や機能を制御しており,病態が増悪しインスリン分泌が低下することがβ細胞のインスリンシグナルを低下させ,さらなるβ細胞の量や機能を障害するという悪循環を惹起していること,また,膵β細胞のPI3キナーゼはその量だけではなく,エキソサイトーシスあるいは膵β細胞どうしのCa2+流入の同期を介して膵β細胞の機能を制御していることを明らかにした(図3).この研究で用いた膵β細胞に特異的なPI3キナーゼノックアウトマウスの表現型はインスリン分泌が低下した耐糖能異常,とくに,インスリン初期分泌の障害を呈するなどヒトの2型糖尿病の初期の状態ときわめて類似しており,2型糖尿病の正確な理解に非常に有用であるものと考えられた.この研究により得られた知見から,今後の2型糖尿病への治療戦略として膵β細胞のPI3キナーゼが新規の治療標的タンパク質としてきわめて有望であり,膵β細胞のPI3キナーゼシグナルを上昇させるような薬剤,あるいは,膵β細胞のインスリン抵抗性を改善するような薬剤が新しい可能性を秘めていることが示唆された.
文 献
- Kulkarni, R.N., Bruning, J. C., Winnay, J. N. et al.: Tissue-specific knockout of the insulin receptor in pancreatic β cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell, 96, 329-339 (1999)[PubMed]
- Ueki, K., Okada, T., Hu, J. et al.: Total insulin and IGF-I resistance in pancreaticβcells causes overt diabetes. Nat. Genet., 38, 583-588 (2006)[PubMed]
- Kubota, N., Terauchi, Y., Tobe, K. et al.: Insulin receptor substrate 2 plays a crucial role in β cells and the hypothalamus. J. Clin. Invest., 114, 917-927 (2004)[PubMed]
- Bernal-Mizrachi, E., Fatrai, S., Johnson, J. D. et al.: Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet βcells. J. Clin. Invest., 114, 928-936 (2004)[PubMed]
- Taniguchi, C. M., Kondo, T., Sajan, M. et al.: Divergent regulation of hepatic glucose and lipid metabolism by phosphoinositide 3-kinase via Akt and PKCλ/ζ. Cell Metab., 3, 343-353 (2006)[PubMed]
- Luo, J., Sobkiw, C. L., Hirshman, M. F. et al.: Loss of class IA PI3K signaling in muscle leads to impaired muscle growth, insulin response, and hyperlipidemia. Cell Metab., 3, 355-366 (2006)[PubMed]
- Rhodes, C. J.: Type 2 diabetes-a matter of β-cell life and death? Science, 307, 380-384 (2005)[PubMed]
- Taniguchi, C. M., Emanuelli, B. & Kahn, C. R.: Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell Biol., 7, 85-96 (2006)[PubMed]
- Takahashi, N., Kishimoto, T., Nemoto, T. et al.: Fusion pore dynamics and insulin granule exocytosis in the pancreatic islet. Science, 297, 1349-1352 (2002)[PubMed]
- Ravier, M., Guldenagel, M., Charollais, A. et al.: Loss of connexin36 channels alters β-cell coupling, islet synchronization of glucose-induced Ca2+ and insulin oscillations, and basal insulin release. Diabetes, 54, 1798-1807 (2005)[PubMed]
著者プロフィール
略歴:2010年 東京大学大学院医学系研究科博士課程 修了,同年より東京大学医学部附属病院 特任研究員.
研究テーマ:膵β細胞におけるPI3キナーゼの役割.
関心事・抱負:2型糖尿病の病態を正確に理解し,それにもとづいた糖尿病の治療薬が開発されるよう,研究に邁進したいと思います.
植木 浩二郎(Kohjiro Ueki)
東京大学大学院医学系研究科 准教授.
© 2010 金子和真・植木浩二郎 Licensed under CC 表示 2.1 日本
