Lkb1は造血幹細胞の細胞周期とエネルギー代謝を制御する
中田 大介
(米国Michigan大学,Center for Stem Cell Biology,Life Sciences Institute,Department of Internal Medicine)
email:中田大介
DOI: 10.7875/first.author.2010.065
Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells.
Daisuke Nakada, Thomas L. Saunders & Sean J. Morrison
Nature, 468, 653-658 (2010)
幹細胞におけるエネルギーの代謝制御およびその分子機構がどのように組織恒常性やがん抑制にかかわっているかについてはほとんど解明されていない.この因果関係を解明するため,筆者らは,エネルギー代謝と細胞成長とを結びつけるがん抑制タンパク質Lkb1とその基質であるAMPキナーゼの解析を行った.Lkb1遺伝子の欠損は造血幹細胞の一時的な増殖とそれにつづく急激な造血幹細胞の枯渇および汎血球減少症をひき起こした.造血幹細胞はLkb1遺伝子の欠損ののち,細胞周期制御や細胞生存の異常をほかの造血未分化細胞より早くに示した.造血幹細胞の枯渇はmTORの活性化や活性酸素種の亢進によるものではなかった.Lkb1遺伝子を欠損した造血幹細胞ではミトコンドリア膜電位差と細胞内ATP量が低下していたが,このことはLkb1遺伝子を欠損したほかの造血未分化細胞ではみられなかった.AMPキナーゼαサブユニットの遺伝子を欠損した造血幹細胞も同様のミトコンドリア機能の異常を示したが,骨髄再構築能は正常であった.Lkb1遺伝子を欠損した造血幹細胞は中心体と紡錘体の異常および染色体異数性を示したが,AMPキナーゼαサブユニットの遺伝子を欠損した造血幹細胞はこれらの異常を示さなかった.これらの結果から,Lkb1はAMPキナーゼに依存的な経路と非依存的な経路とを介して造血幹細胞を制御していることが明らかになった.また,造血幹細胞とほかの造血未分化細胞ではエネルギー代謝と細胞周期の制御機構が大きく異なっていることがうきぼりとなった.
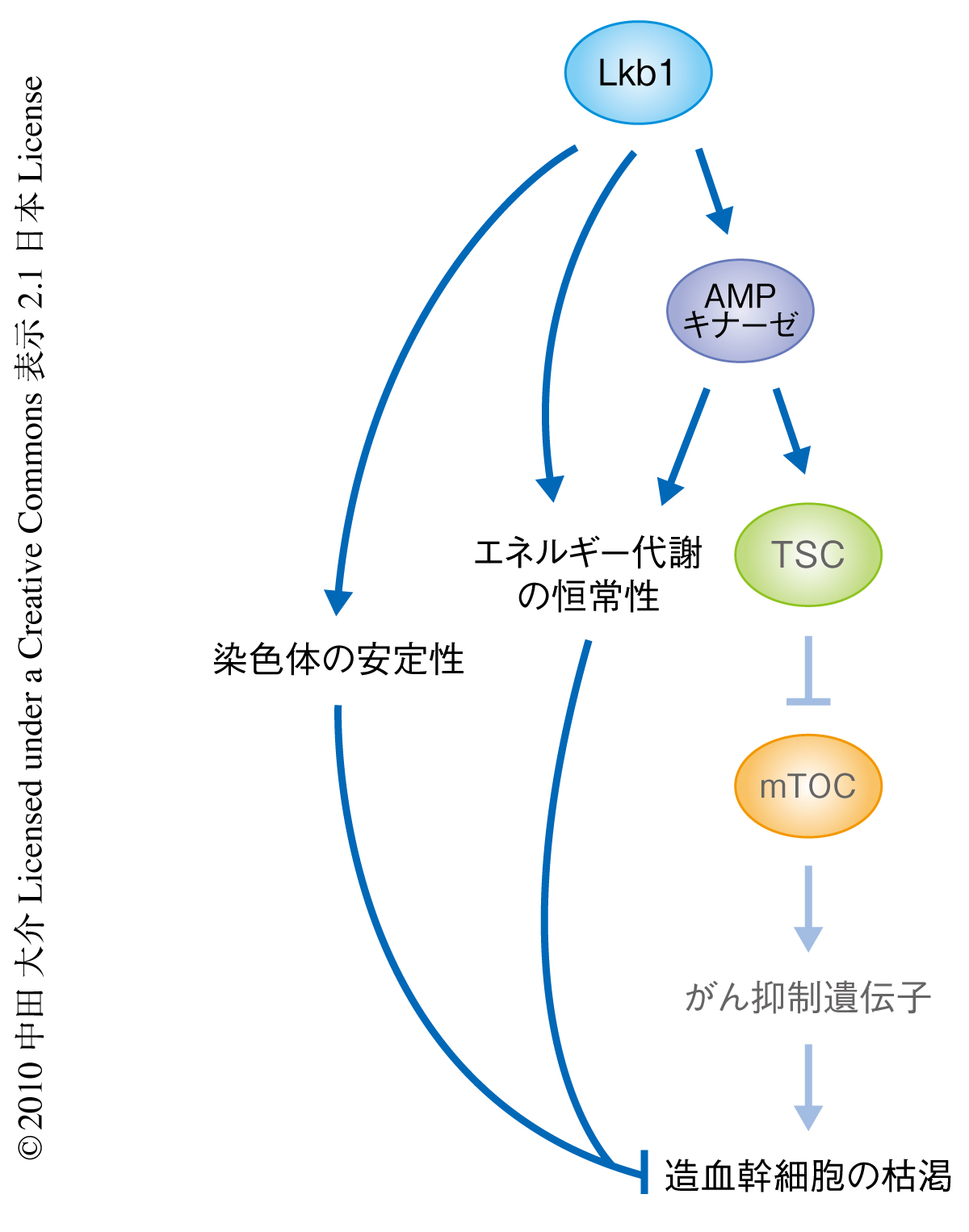
Lkb1は細胞成長とエネルギー代謝とを結びつける重要な経路を構成している1).エネルギーの枯渇によるストレスはLkb1を介して異化反応やミトコンドリアの合成を促し,また,mTORによるタンパク質合成を含む同化反応の亢進を抑制している.Lkb1はAMPキナーゼとAMPキナーゼ様キナーゼを介してこれらの経路を制御している.AMPキナーゼは結節性硬化症の原因タンパク質であるTSCを活性化し,TSCはmTORを抑制することでタンパク質合成や細胞成長を抑制している2).AMPキナーゼはRaptorをリン酸化することによりmTORを抑制することもできる3).また,AMPキナーゼは転写因子FoxOを活性化し,エネルギー代謝や細胞周期,アポトーシス,抗酸化ストレス応答を制御している4).
Lkb1は胚発生と成体の分化した細胞での代謝と極性を制御している.線虫C. elegansのLkb1ホモログは胚の極性を決定し,ショウジョウバエのLkb1ホモログおよびAMPキナーゼホモログは胚の細胞極性,非対称分裂,紡垂体形成を制御している.Lkb1ノックアウトマウスは血管形成や神経管に異常をきたし妊娠中期に死亡する5,6).また,Lkb1は成体の筋組織,肝臓,膵臓,T細胞のエネルギー代謝を制御している.Lkb1遺伝子の欠損は神経,上皮細胞,膵臓β細胞の極性や分化に異常をもたらす.しかし,Lkb1が幹細胞の維持や組織恒常性の維持にかかわっているのかどうかは不明である.
Lkb1遺伝子の欠損はさまざまな細胞の増殖を亢進させることが知られている.Lkb1遺伝子を欠損したマウス胚線維芽細胞は不死化し7),高頻度に上皮細胞腫瘍を発症するPeutz-Jeghers症候群においてLkb1遺伝子の変異がみつかっている1).これらの結果は,Lkb1の主要な機能は細胞の異常増殖,ひいては,組織の過成長を抑えることであることを示唆している.Lkb1が幹細胞の機能をどのように制御しているのかを解明するため,筆者らは,Lkb1遺伝子を造血細胞から欠損させる解析を行った.
まず,造血未分化細胞においてLkb1遺伝子の発現を解析したところ,造血幹細胞画分や8),短期的に骨髄を再構築する系譜を制限された未分化細胞(restricted progenitor,造血幹細胞より分化しているが,顆粒球・マクロファージ前駆体より未分化な細胞)の画分,顆粒球・マクロファージ前駆体画分は,全骨髄細胞画分と比較して,2倍ほどLkb1遺伝子の発現の高いことを見い出した.
つぎに,Lkb1遺伝子を誘導的に造血細胞から欠損させることのできるコンディショナルノックアウトマウスを作製した.Lkb1遺伝子の欠損6日後から18日後では造血組織の細胞数に大きな影響はみられなかったが,欠損24日後から34日後において汎血球減少症が認められた.Lkb1遺伝子の欠損2日後から6日後では造血幹細胞画分の細胞数が2倍ほどに増加したが,欠損18日後には対照群の1/7にまで低下していた.これらの結果から,Lkb1遺伝子の欠損は造血幹細胞の枯渇とそれにつづく汎血球減少症をひき起こすことがわかった.
つづいて,Lkb1は造血細胞の細胞周期の進行を制御しているのかどうか検討した.Lkb1遺伝子の欠損6日後では造血幹細胞画分と系譜を制限された未分化細胞画分では細胞周期進行の指標となるBrdUの取り込みが大きく亢進していた.これらの未分化細胞では細胞周期のG1期やS期/G2期/M期にいる細胞の割合が上昇していた.これらの変化は顆粒球・マクロファージ前駆体細胞画分や全骨髄細胞画分ではみられなかった.つまり,Lkb1は造血幹細胞の細胞周期の進行を抑えていることがわかった.
さらに,Lkb1が造血細胞の細胞死を制御しているのかどうか検討した.Lkb1遺伝子の欠損11日後に造血幹細胞画分においてアポトーシスを亢進するカスパーゼの活性が亢進した.この時点では,ほかの造血未分化細胞ではカスパーゼ活性の亢進は認められなかった.ただし,Lkb1遺伝子の欠損24日後では多くの造血細胞でカスパーゼ活性の亢進がみられた.これらの結果から,造血幹細胞はLkb1遺伝子の欠損ののち,ほかの造血未分化細胞と比べてより急速に細胞死の誘導されることが明らかになった.
つぎに,Lkb1遺伝子を欠損した造血幹細胞画分の長期の骨髄再構築能を競合的な骨髄移植により評価したところ,骨髄再構築能を著しく欠くことがわかった.この再構築能の異常が細胞自立的であるかどうかを検討するため,Lkb1遺伝子の誘導的な欠損を起こさせるまえのマウスから骨髄細胞を採取し,これを移植したマウスで誘導的な欠損を起こさせることで,移植された骨髄細胞において特異的にLkb1遺伝子を欠損させた.Lkb1遺伝子の欠損を誘導したのち移植された骨髄細胞に由来する血液細胞は急速に減少した.さらに,Lkb1遺伝子の欠損誘導の2ヵ月後にはLkb1遺伝子を欠損した造血幹細胞画分は骨髄において枯渇していることを見い出した.これらの結果から,Lkb1は細胞自立的に造血幹細胞の骨髄再構築能を制御していることが明らかになった.
Lkb1遺伝子は造血幹細胞画分が培養下でコロニーを形成するのにも必須であった.Lkb1遺伝子を欠損した造血幹細胞画分のコロニー形成能は著しく低く,また,形成されたコロニーは著しく小さいものであった.Lkb1遺伝子を欠損した全骨髄細胞画分もコロニー形成能の低下を示した.ただし,Lkb1遺伝子を欠損した顆粒球・マクロファージ前駆体画分のコロニー形成能は正常であったことから,すべての未分化細胞がコロニー形成能に異常をきたすわけではないことが示された.
Lkb1遺伝子の欠損によりAMPキナーゼが不活性化しているのかどうかを確認するため,造血幹細胞画分と,系譜を制限された未分化細胞画分,顆粒球・マクロファージ前駆体画分,全骨髄細胞画分を,Lkb1遺伝子の欠損誘導から6日後に分取しウェスタンブロット解析を行った.Lkb1はすべての画分で発現がみられ,Lkb1遺伝子の欠損によりその発現は消失した.Lkb1によるリン酸化部位であるAMPキナーゼαサブユニットの172番目のスレオニン残基のリン酸化はLkb1遺伝子を欠損した造血幹細胞画分と系譜を制限された未分化細胞画分で減少していたが,ほかの画分では影響はみられなかった.AMPキナーゼの基質であるACCのリン酸化は造血幹細胞画分で減少していたが,ほかの画分では影響はみられなかった.Lkb1遺伝子の欠損誘導から24日後に全骨髄細胞画分を用いてウェスタンブロット解析を行ったところ,AMPキナーゼαサブユニットとACCのリン酸化は大きく減少していた.これらの結果から,Lkb1はさまざまな血液細胞においてAMPキナーゼのリン酸化を制御しているが,造血幹細胞はそのなかでもとくに急速にLkb1遺伝子の欠損の影響をうけ,AMPキナーゼ経路の不活性化の起こるものと考えられた.
AMPキナーゼはmTORシグナルを抑制し,mTORシグナルの亢進は造血幹細胞の枯渇をまねくことから9,10),Lkb1遺伝子を欠損した造血幹細胞画分においてmTORシグナルが亢進しているかどうか,S6や4EBPのリン酸化の解析により検討した.その結果,S6と4EBPのリン酸化はLkb1遺伝子を欠損した系譜を制限された未分化細胞画分や顆粒球・マクロファージ前駆体画分,全骨髄細胞画分で亢進がみられたが,造血幹細胞画分では影響はみられなかった.これらの結果から,Lkb1によるmTORシグナルの制御は造血幹細胞とほかの造血未分化細胞とで異なることが示唆された.
Lkb1遺伝子を欠損した造血幹細胞画分の枯渇にmTORシグナルの亢進がかかわっているかどうか機能的に検討するため,mTOR阻害剤であるラパマイシンが造血幹細胞画分の枯渇を抑制できるかどうかを解析した.その結果,Lkb1遺伝子の欠損誘導ののち1ヵ月のあいだラパマイシンを投与しても造血幹細胞画分は枯渇してしまうことを見い出した.また,Lkb1遺伝子を誘導的に欠損するまえの骨髄細胞を移植したマウスにLkb1遺伝子の誘導的な欠損を起こさせたのち,ラパマイシンを投与して移植ののちの造血幹細胞画分の枯渇を抑制できるかどうか検討したが,やはり,ラパマイシンは造血幹細胞画分の枯渇を防ぐことはできなかった.以上の結果から,Lkb1遺伝子を欠損したのちの造血幹細胞の枯渇はmTORシグナルとは異なる分子機構によるものであることが強く示唆された.
AMPキナーゼαサブユニットをコードするPrkaa1遺伝子とPrkaa2遺伝子の発現を血液細胞で解析したところ,両者ともほかの造血未分化細胞と比べ造血幹細胞画分で発現量の高いことを見い出した.AMPキナーゼの造血幹細胞における機能を解析するため,この2つの遺伝子を誘導的に造血細胞から欠損させることのできるコンディショナルノックアウトマウスを作製した.AMPキナーゼαサブユニットの遺伝子を欠損した造血幹細胞画分,系譜を制限された未分化細胞画分,顆粒球・マクロファージ前駆体画分,全骨髄細胞画分では,AMPキナーゼαサブユニットの発現とリン酸化,また,ACCのリン酸化が大きく減少していた.また,mTORシグナルの指標であるS6のリン酸化はどの画分でも上昇していた.これらの結果から,AMPキナーゼはさまざまな血液細胞においてmTORシグナルを抑制していることが明らかとなった.
転写因子FoxOをコードする遺伝子を欠損した造血幹細胞は活性酸素種の亢進により枯渇し,AMPキナーゼはFoxOを活性化することのできることから,Lkb1遺伝子やAMPキナーゼαサブユニットの遺伝子を欠損した造血幹細胞画分では活性酸素種が亢進しているのかどうかを検討した.その結果,活性酸素種の亢進はみられず,また,抗酸化剤であるNAC(N-アセチル-L-システイン)の投与はLkb1遺伝子を欠損した造血幹細胞画分の枯渇を抑制することはできなかった.
つぎに,Lkb1遺伝子やAMPキナーゼαサブユニットの遺伝子を欠損した造血幹細胞画分におけるミトコンドリアの機能を解析した.その結果,Lkb1遺伝子やAMPキナーゼαサブユニットの遺伝子を欠損した造血幹細胞画分ではミトコンドリアの細胞内容積の異常な亢進がみられた.これには,Lkb1-AMPキナーゼ経路によるミトコンドリアの過大化の抑制,もしくは,ミトコンドリアの機能不全や細胞内ATP量の減少によるフィードバック機構による可能性が考えられた.実際,Tfam遺伝子の欠損によるミトコンドリアの機能不全と過大化が報告されている.後者の可能性と一致して,Lkb1遺伝子やAMPキナーゼαサブユニットの遺伝子を欠損した造血幹細胞画分ではミトコンドリアDNAのコピー数の減少がみられた.また,Lkb1遺伝子を欠損した造血幹細胞画分ではミトコンドリア膜電位の減少がみられた.このミトコンドリア膜電位の減少は顆粒球・マクロファージ前駆体画分や全骨髄細胞画分では認められなかった.以上の結果から,Lkb1はAMPキナーゼを介した経路とAMPキナーゼを介さない経路とによりミトコンドリアの機能を制御していることがわかった.
さらに,造血幹細胞画分の細胞内ATP量を解析した.ここで,死細胞や骨髄内の細胞外基質などがATP量の検定に影響をあたえることを防ぐため,造血幹細胞画分やほかの血液細胞画分をセルソーターにより分取してATP量を細胞数により標準化した.解析の結果,Lkb1遺伝子やAMPキナーゼαサブユニットの遺伝子を欠損した造血幹細胞画分ではATP量の減少がみられた.つまり,Lkb1-AMPキナーゼ経路は造血幹細胞のミトコンドリア機能とエネルギー代謝の恒常性を制御していることが明らかになった.
AMPキナーゼαサブユニットの遺伝子の欠損はミトコンドリア機能の制御などLkb1遺伝子の欠損と同様の異常をもたらしたが,造血幹細胞の維持においては必須ではないことを見い出した.Lkb1遺伝子を欠損したのちにみられた造血幹細胞画分の一時的な増加とそれにつづく枯渇は,AMPキナーゼαサブユニットの遺伝子を欠損したあとにはみられなかった.また,AMPキナーゼαサブユニットの遺伝子を欠損した造血幹細胞画分は移植ののちの骨髄再構築能を正常にもちあわせていることも示された.これらの結果から,Lkb1はおもにAMPキナーゼに非依存的な機構により造血幹細胞の維持制御を行っているものと考えられた.
Lkb1遺伝子を欠損した造血幹細胞の分裂を詳細に解析するため,造血幹細胞画分をその機能の維持できる培養液に播種してin vitroにおける増殖を観察した.野生型の造血幹細胞画分とLkb1遺伝子を欠損した造血幹細胞画分は3日間の培養でほぼすべての細胞が分裂した.野生型の造血幹細胞画分はそののち対数的に増殖したのに対し,Lkb1遺伝子を欠損した造血幹細胞画分はそののちほとんど分裂しなかった.Lkb1遺伝子を欠損した造血幹細胞画分はBrdUの正常な取り込みを示したため細胞分裂能の異常はS期進入の異常によるものではないと考えられた.一方で,Lkb1遺伝子を欠損した造血幹細胞画分はM期のマーカーであるリン酸化型ヒストンH3陽性の細胞数が減少していたことから,M期進入もしくはM期進行の異常を示しているものと考えられた.
驚くべきことに,多くのLkb1遺伝子を欠損した造血幹細胞画分は中心体の異数性および紡錘体の形成異常を示した.この異常は野生型の造血幹細胞画分やLkb1遺伝子を欠損した顆粒球・マクロファージ前駆体画分ではみられなかった.この分裂機構の異常が細胞死をまねくのかどうか検討したところ,Lkb1遺伝子を欠損した造血幹細胞画分の多くの細胞は細胞死のマーカーであるアネキシンVに陽性を示した.さらに,Lkb1遺伝子を欠損した造血幹細胞画分は染色体の異数性も示した.Lkb1遺伝子を欠損した顆粒球・マクロファージ前駆体画分では染色体の異数性はみられなかった.また,AMPキナーゼαサブユニットの遺伝子を欠損した造血幹細胞画分も染色体の異数性を示さなかった.以上の結果から,Lkb1はAMPキナーゼ非依存的に造血幹細胞の染色体の正常な分配を制御していることが明らかになった.
今回の研究により,Lkb1遺伝子の欠損は一時的な造血幹細胞の増殖とそれにつづく急速な枯渇をまねき,汎血球減少症をひき起こすことが明らかとなった(図1).Lkb1はmTORや活性酸素種を介さない分子機構により造血幹細胞を制御していた.Lkb1遺伝子やAMPキナーゼα遺伝子の欠損は造血幹細胞のミトコンドリア機能とエネルギー代謝の恒常性を阻害したが,AMPキナーゼαサブユニットの遺伝子の欠損が造血幹細胞の機能にあたえた影響はごくわずかであった.Lkb1はAMPキナーゼ非依存的に細胞分裂や染色体分配を制御していたことから,染色体分配の機能異常が造血幹細胞の機能の枯渇をまねいた可能性が考えられた.しかし,Lkb1がどのように染色体の異数性を抑制しているかはいまだ不明であり,M期進入やM期進行の異常などによる二次的な影響により染色体分配が破綻した可能性も考えられる.今後の研究により,Lkb1がどのように染色体の異数性を抑制しているかを明らかにしていきたい.
略歴:2005年 名古屋大学大学院理学研究科 修了,2006年より日本学術振興会 海外特別研究員(米国Michigan大学Sean Morrison研究室).
研究テーマ:幹細胞の自己複製を制御する分子機構の解明.
抱負:分子生物学的な手法,遺伝学的な手法を用いて,幹細胞をつかさどる難解な分子機構を解き明かしたい.
© 2010 中田 大介 Licensed under CC 表示 2.1 日本
(米国Michigan大学,Center for Stem Cell Biology,Life Sciences Institute,Department of Internal Medicine)
email:中田大介
DOI: 10.7875/first.author.2010.065
Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells.
Daisuke Nakada, Thomas L. Saunders & Sean J. Morrison
Nature, 468, 653-658 (2010)
要 約
幹細胞におけるエネルギーの代謝制御およびその分子機構がどのように組織恒常性やがん抑制にかかわっているかについてはほとんど解明されていない.この因果関係を解明するため,筆者らは,エネルギー代謝と細胞成長とを結びつけるがん抑制タンパク質Lkb1とその基質であるAMPキナーゼの解析を行った.Lkb1遺伝子の欠損は造血幹細胞の一時的な増殖とそれにつづく急激な造血幹細胞の枯渇および汎血球減少症をひき起こした.造血幹細胞はLkb1遺伝子の欠損ののち,細胞周期制御や細胞生存の異常をほかの造血未分化細胞より早くに示した.造血幹細胞の枯渇はmTORの活性化や活性酸素種の亢進によるものではなかった.Lkb1遺伝子を欠損した造血幹細胞ではミトコンドリア膜電位差と細胞内ATP量が低下していたが,このことはLkb1遺伝子を欠損したほかの造血未分化細胞ではみられなかった.AMPキナーゼαサブユニットの遺伝子を欠損した造血幹細胞も同様のミトコンドリア機能の異常を示したが,骨髄再構築能は正常であった.Lkb1遺伝子を欠損した造血幹細胞は中心体と紡錘体の異常および染色体異数性を示したが,AMPキナーゼαサブユニットの遺伝子を欠損した造血幹細胞はこれらの異常を示さなかった.これらの結果から,Lkb1はAMPキナーゼに依存的な経路と非依存的な経路とを介して造血幹細胞を制御していることが明らかになった.また,造血幹細胞とほかの造血未分化細胞ではエネルギー代謝と細胞周期の制御機構が大きく異なっていることがうきぼりとなった.
はじめに
Lkb1は細胞成長とエネルギー代謝とを結びつける重要な経路を構成している1).エネルギーの枯渇によるストレスはLkb1を介して異化反応やミトコンドリアの合成を促し,また,mTORによるタンパク質合成を含む同化反応の亢進を抑制している.Lkb1はAMPキナーゼとAMPキナーゼ様キナーゼを介してこれらの経路を制御している.AMPキナーゼは結節性硬化症の原因タンパク質であるTSCを活性化し,TSCはmTORを抑制することでタンパク質合成や細胞成長を抑制している2).AMPキナーゼはRaptorをリン酸化することによりmTORを抑制することもできる3).また,AMPキナーゼは転写因子FoxOを活性化し,エネルギー代謝や細胞周期,アポトーシス,抗酸化ストレス応答を制御している4).
Lkb1は胚発生と成体の分化した細胞での代謝と極性を制御している.線虫C. elegansのLkb1ホモログは胚の極性を決定し,ショウジョウバエのLkb1ホモログおよびAMPキナーゼホモログは胚の細胞極性,非対称分裂,紡垂体形成を制御している.Lkb1ノックアウトマウスは血管形成や神経管に異常をきたし妊娠中期に死亡する5,6).また,Lkb1は成体の筋組織,肝臓,膵臓,T細胞のエネルギー代謝を制御している.Lkb1遺伝子の欠損は神経,上皮細胞,膵臓β細胞の極性や分化に異常をもたらす.しかし,Lkb1が幹細胞の維持や組織恒常性の維持にかかわっているのかどうかは不明である.
Lkb1遺伝子の欠損はさまざまな細胞の増殖を亢進させることが知られている.Lkb1遺伝子を欠損したマウス胚線維芽細胞は不死化し7),高頻度に上皮細胞腫瘍を発症するPeutz-Jeghers症候群においてLkb1遺伝子の変異がみつかっている1).これらの結果は,Lkb1の主要な機能は細胞の異常増殖,ひいては,組織の過成長を抑えることであることを示唆している.Lkb1が幹細胞の機能をどのように制御しているのかを解明するため,筆者らは,Lkb1遺伝子を造血細胞から欠損させる解析を行った.
1.Lkb1遺伝子の欠損は造血幹細胞を枯渇させる
まず,造血未分化細胞においてLkb1遺伝子の発現を解析したところ,造血幹細胞画分や8),短期的に骨髄を再構築する系譜を制限された未分化細胞(restricted progenitor,造血幹細胞より分化しているが,顆粒球・マクロファージ前駆体より未分化な細胞)の画分,顆粒球・マクロファージ前駆体画分は,全骨髄細胞画分と比較して,2倍ほどLkb1遺伝子の発現の高いことを見い出した.
つぎに,Lkb1遺伝子を誘導的に造血細胞から欠損させることのできるコンディショナルノックアウトマウスを作製した.Lkb1遺伝子の欠損6日後から18日後では造血組織の細胞数に大きな影響はみられなかったが,欠損24日後から34日後において汎血球減少症が認められた.Lkb1遺伝子の欠損2日後から6日後では造血幹細胞画分の細胞数が2倍ほどに増加したが,欠損18日後には対照群の1/7にまで低下していた.これらの結果から,Lkb1遺伝子の欠損は造血幹細胞の枯渇とそれにつづく汎血球減少症をひき起こすことがわかった.
つづいて,Lkb1は造血細胞の細胞周期の進行を制御しているのかどうか検討した.Lkb1遺伝子の欠損6日後では造血幹細胞画分と系譜を制限された未分化細胞画分では細胞周期進行の指標となるBrdUの取り込みが大きく亢進していた.これらの未分化細胞では細胞周期のG1期やS期/G2期/M期にいる細胞の割合が上昇していた.これらの変化は顆粒球・マクロファージ前駆体細胞画分や全骨髄細胞画分ではみられなかった.つまり,Lkb1は造血幹細胞の細胞周期の進行を抑えていることがわかった.
さらに,Lkb1が造血細胞の細胞死を制御しているのかどうか検討した.Lkb1遺伝子の欠損11日後に造血幹細胞画分においてアポトーシスを亢進するカスパーゼの活性が亢進した.この時点では,ほかの造血未分化細胞ではカスパーゼ活性の亢進は認められなかった.ただし,Lkb1遺伝子の欠損24日後では多くの造血細胞でカスパーゼ活性の亢進がみられた.これらの結果から,造血幹細胞はLkb1遺伝子の欠損ののち,ほかの造血未分化細胞と比べてより急速に細胞死の誘導されることが明らかになった.
2.Lkb1遺伝子を欠損した造血幹細胞は長期の骨髄再構築能を欠く
つぎに,Lkb1遺伝子を欠損した造血幹細胞画分の長期の骨髄再構築能を競合的な骨髄移植により評価したところ,骨髄再構築能を著しく欠くことがわかった.この再構築能の異常が細胞自立的であるかどうかを検討するため,Lkb1遺伝子の誘導的な欠損を起こさせるまえのマウスから骨髄細胞を採取し,これを移植したマウスで誘導的な欠損を起こさせることで,移植された骨髄細胞において特異的にLkb1遺伝子を欠損させた.Lkb1遺伝子の欠損を誘導したのち移植された骨髄細胞に由来する血液細胞は急速に減少した.さらに,Lkb1遺伝子の欠損誘導の2ヵ月後にはLkb1遺伝子を欠損した造血幹細胞画分は骨髄において枯渇していることを見い出した.これらの結果から,Lkb1は細胞自立的に造血幹細胞の骨髄再構築能を制御していることが明らかになった.
Lkb1遺伝子は造血幹細胞画分が培養下でコロニーを形成するのにも必須であった.Lkb1遺伝子を欠損した造血幹細胞画分のコロニー形成能は著しく低く,また,形成されたコロニーは著しく小さいものであった.Lkb1遺伝子を欠損した全骨髄細胞画分もコロニー形成能の低下を示した.ただし,Lkb1遺伝子を欠損した顆粒球・マクロファージ前駆体画分のコロニー形成能は正常であったことから,すべての未分化細胞がコロニー形成能に異常をきたすわけではないことが示された.
3.Lkb1遺伝子を欠損した造血幹細胞はmTOR非依存的に枯渇する
Lkb1遺伝子の欠損によりAMPキナーゼが不活性化しているのかどうかを確認するため,造血幹細胞画分と,系譜を制限された未分化細胞画分,顆粒球・マクロファージ前駆体画分,全骨髄細胞画分を,Lkb1遺伝子の欠損誘導から6日後に分取しウェスタンブロット解析を行った.Lkb1はすべての画分で発現がみられ,Lkb1遺伝子の欠損によりその発現は消失した.Lkb1によるリン酸化部位であるAMPキナーゼαサブユニットの172番目のスレオニン残基のリン酸化はLkb1遺伝子を欠損した造血幹細胞画分と系譜を制限された未分化細胞画分で減少していたが,ほかの画分では影響はみられなかった.AMPキナーゼの基質であるACCのリン酸化は造血幹細胞画分で減少していたが,ほかの画分では影響はみられなかった.Lkb1遺伝子の欠損誘導から24日後に全骨髄細胞画分を用いてウェスタンブロット解析を行ったところ,AMPキナーゼαサブユニットとACCのリン酸化は大きく減少していた.これらの結果から,Lkb1はさまざまな血液細胞においてAMPキナーゼのリン酸化を制御しているが,造血幹細胞はそのなかでもとくに急速にLkb1遺伝子の欠損の影響をうけ,AMPキナーゼ経路の不活性化の起こるものと考えられた.
AMPキナーゼはmTORシグナルを抑制し,mTORシグナルの亢進は造血幹細胞の枯渇をまねくことから9,10),Lkb1遺伝子を欠損した造血幹細胞画分においてmTORシグナルが亢進しているかどうか,S6や4EBPのリン酸化の解析により検討した.その結果,S6と4EBPのリン酸化はLkb1遺伝子を欠損した系譜を制限された未分化細胞画分や顆粒球・マクロファージ前駆体画分,全骨髄細胞画分で亢進がみられたが,造血幹細胞画分では影響はみられなかった.これらの結果から,Lkb1によるmTORシグナルの制御は造血幹細胞とほかの造血未分化細胞とで異なることが示唆された.
Lkb1遺伝子を欠損した造血幹細胞画分の枯渇にmTORシグナルの亢進がかかわっているかどうか機能的に検討するため,mTOR阻害剤であるラパマイシンが造血幹細胞画分の枯渇を抑制できるかどうかを解析した.その結果,Lkb1遺伝子の欠損誘導ののち1ヵ月のあいだラパマイシンを投与しても造血幹細胞画分は枯渇してしまうことを見い出した.また,Lkb1遺伝子を誘導的に欠損するまえの骨髄細胞を移植したマウスにLkb1遺伝子の誘導的な欠損を起こさせたのち,ラパマイシンを投与して移植ののちの造血幹細胞画分の枯渇を抑制できるかどうか検討したが,やはり,ラパマイシンは造血幹細胞画分の枯渇を防ぐことはできなかった.以上の結果から,Lkb1遺伝子を欠損したのちの造血幹細胞の枯渇はmTORシグナルとは異なる分子機構によるものであることが強く示唆された.
4.Lkb1は造血幹細胞のミトコンドリア機能を制御する
AMPキナーゼαサブユニットをコードするPrkaa1遺伝子とPrkaa2遺伝子の発現を血液細胞で解析したところ,両者ともほかの造血未分化細胞と比べ造血幹細胞画分で発現量の高いことを見い出した.AMPキナーゼの造血幹細胞における機能を解析するため,この2つの遺伝子を誘導的に造血細胞から欠損させることのできるコンディショナルノックアウトマウスを作製した.AMPキナーゼαサブユニットの遺伝子を欠損した造血幹細胞画分,系譜を制限された未分化細胞画分,顆粒球・マクロファージ前駆体画分,全骨髄細胞画分では,AMPキナーゼαサブユニットの発現とリン酸化,また,ACCのリン酸化が大きく減少していた.また,mTORシグナルの指標であるS6のリン酸化はどの画分でも上昇していた.これらの結果から,AMPキナーゼはさまざまな血液細胞においてmTORシグナルを抑制していることが明らかとなった.
転写因子FoxOをコードする遺伝子を欠損した造血幹細胞は活性酸素種の亢進により枯渇し,AMPキナーゼはFoxOを活性化することのできることから,Lkb1遺伝子やAMPキナーゼαサブユニットの遺伝子を欠損した造血幹細胞画分では活性酸素種が亢進しているのかどうかを検討した.その結果,活性酸素種の亢進はみられず,また,抗酸化剤であるNAC(N-アセチル-L-システイン)の投与はLkb1遺伝子を欠損した造血幹細胞画分の枯渇を抑制することはできなかった.
つぎに,Lkb1遺伝子やAMPキナーゼαサブユニットの遺伝子を欠損した造血幹細胞画分におけるミトコンドリアの機能を解析した.その結果,Lkb1遺伝子やAMPキナーゼαサブユニットの遺伝子を欠損した造血幹細胞画分ではミトコンドリアの細胞内容積の異常な亢進がみられた.これには,Lkb1-AMPキナーゼ経路によるミトコンドリアの過大化の抑制,もしくは,ミトコンドリアの機能不全や細胞内ATP量の減少によるフィードバック機構による可能性が考えられた.実際,Tfam遺伝子の欠損によるミトコンドリアの機能不全と過大化が報告されている.後者の可能性と一致して,Lkb1遺伝子やAMPキナーゼαサブユニットの遺伝子を欠損した造血幹細胞画分ではミトコンドリアDNAのコピー数の減少がみられた.また,Lkb1遺伝子を欠損した造血幹細胞画分ではミトコンドリア膜電位の減少がみられた.このミトコンドリア膜電位の減少は顆粒球・マクロファージ前駆体画分や全骨髄細胞画分では認められなかった.以上の結果から,Lkb1はAMPキナーゼを介した経路とAMPキナーゼを介さない経路とによりミトコンドリアの機能を制御していることがわかった.
さらに,造血幹細胞画分の細胞内ATP量を解析した.ここで,死細胞や骨髄内の細胞外基質などがATP量の検定に影響をあたえることを防ぐため,造血幹細胞画分やほかの血液細胞画分をセルソーターにより分取してATP量を細胞数により標準化した.解析の結果,Lkb1遺伝子やAMPキナーゼαサブユニットの遺伝子を欠損した造血幹細胞画分ではATP量の減少がみられた.つまり,Lkb1-AMPキナーゼ経路は造血幹細胞のミトコンドリア機能とエネルギー代謝の恒常性を制御していることが明らかになった.
AMPキナーゼαサブユニットの遺伝子の欠損はミトコンドリア機能の制御などLkb1遺伝子の欠損と同様の異常をもたらしたが,造血幹細胞の維持においては必須ではないことを見い出した.Lkb1遺伝子を欠損したのちにみられた造血幹細胞画分の一時的な増加とそれにつづく枯渇は,AMPキナーゼαサブユニットの遺伝子を欠損したあとにはみられなかった.また,AMPキナーゼαサブユニットの遺伝子を欠損した造血幹細胞画分は移植ののちの骨髄再構築能を正常にもちあわせていることも示された.これらの結果から,Lkb1はおもにAMPキナーゼに非依存的な機構により造血幹細胞の維持制御を行っているものと考えられた.
5.Lkb1遺伝子を欠損した造血幹細胞は染色体の異数性を示す
Lkb1遺伝子を欠損した造血幹細胞の分裂を詳細に解析するため,造血幹細胞画分をその機能の維持できる培養液に播種してin vitroにおける増殖を観察した.野生型の造血幹細胞画分とLkb1遺伝子を欠損した造血幹細胞画分は3日間の培養でほぼすべての細胞が分裂した.野生型の造血幹細胞画分はそののち対数的に増殖したのに対し,Lkb1遺伝子を欠損した造血幹細胞画分はそののちほとんど分裂しなかった.Lkb1遺伝子を欠損した造血幹細胞画分はBrdUの正常な取り込みを示したため細胞分裂能の異常はS期進入の異常によるものではないと考えられた.一方で,Lkb1遺伝子を欠損した造血幹細胞画分はM期のマーカーであるリン酸化型ヒストンH3陽性の細胞数が減少していたことから,M期進入もしくはM期進行の異常を示しているものと考えられた.
驚くべきことに,多くのLkb1遺伝子を欠損した造血幹細胞画分は中心体の異数性および紡錘体の形成異常を示した.この異常は野生型の造血幹細胞画分やLkb1遺伝子を欠損した顆粒球・マクロファージ前駆体画分ではみられなかった.この分裂機構の異常が細胞死をまねくのかどうか検討したところ,Lkb1遺伝子を欠損した造血幹細胞画分の多くの細胞は細胞死のマーカーであるアネキシンVに陽性を示した.さらに,Lkb1遺伝子を欠損した造血幹細胞画分は染色体の異数性も示した.Lkb1遺伝子を欠損した顆粒球・マクロファージ前駆体画分では染色体の異数性はみられなかった.また,AMPキナーゼαサブユニットの遺伝子を欠損した造血幹細胞画分も染色体の異数性を示さなかった.以上の結果から,Lkb1はAMPキナーゼ非依存的に造血幹細胞の染色体の正常な分配を制御していることが明らかになった.
おわりに
今回の研究により,Lkb1遺伝子の欠損は一時的な造血幹細胞の増殖とそれにつづく急速な枯渇をまねき,汎血球減少症をひき起こすことが明らかとなった(図1).Lkb1はmTORや活性酸素種を介さない分子機構により造血幹細胞を制御していた.Lkb1遺伝子やAMPキナーゼα遺伝子の欠損は造血幹細胞のミトコンドリア機能とエネルギー代謝の恒常性を阻害したが,AMPキナーゼαサブユニットの遺伝子の欠損が造血幹細胞の機能にあたえた影響はごくわずかであった.Lkb1はAMPキナーゼ非依存的に細胞分裂や染色体分配を制御していたことから,染色体分配の機能異常が造血幹細胞の機能の枯渇をまねいた可能性が考えられた.しかし,Lkb1がどのように染色体の異数性を抑制しているかはいまだ不明であり,M期進入やM期進行の異常などによる二次的な影響により染色体分配が破綻した可能性も考えられる.今後の研究により,Lkb1がどのように染色体の異数性を抑制しているかを明らかにしていきたい.
文 献
- Shackelford, D. B. & Shaw, R. J.: The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat. Rev. Cancer, 9, 563-575 (2009)[PubMed]
- Inoki, K., Zhu, T. & Guan, K. L.: TSC2 mediates cellular energy response to control cell growth and survival. Cell, 115, 577-590 (2003)[PubMed]
- Gwinn, D. M., Shackelford, D. B., Egan, D. F. et al.: AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell, 30, 214-226 (2008)[PubMed]
- Canto, C., Gerhart-Hines, Z., Feige, J. N. et al.: AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature, 458, 1056-1060 (2009)[PubMed]
- Jishage, K., Nezu, J., Kawase, Y. et al.: Role of Lkb1, the causative gene of Peutz-Jegher's syndrome, in embryogenesis and polyposis. Proc. Natl. Acad. Sci. USA, 99, 8903-8908 (2002)[PubMed]
- Ylikorkala, A., Rossi, D. J., Korsisaari, N. et al.: Vascular abnormalities and deregulation of VEGF in Lkb1-deficient mice. Science, 293, 1323-1326 (2001)[PubMed]
- Bardeesy, N., Sinha, M., Hezel, A. F. et al.: Loss of the Lkb1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature, 419, 162-167 (2002)[PubMed]
- Kiel, M. J., Yilmaz, O. H., Iwashita, T. et al.: SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell, 121, 1109-1121 (2005)[PubMed]
- Yilmaz, O. H., Valdez, R., Theisen, B. K. et al.: Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature, 441, 475-482 (2006)[PubMed]
- Lee, J. Y , Nakada, D., Yilmaz, O. H. et al.: mTOR activation induces tumor suppressors that inhibit leukemogenesis and deplete hematopoietic stem cells after Pten deletion. Cell Stem Cell, 7, 593-605 (2010)[PubMed] [新着論文レビュー]
著者プロフィール
略歴:2005年 名古屋大学大学院理学研究科 修了,2006年より日本学術振興会 海外特別研究員(米国Michigan大学Sean Morrison研究室).
研究テーマ:幹細胞の自己複製を制御する分子機構の解明.
抱負:分子生物学的な手法,遺伝学的な手法を用いて,幹細胞をつかさどる難解な分子機構を解き明かしたい.
© 2010 中田 大介 Licensed under CC 表示 2.1 日本
