細胞表面のリガンドSasおよびその受容体PTP10Dはがんの抑制にはたらく細胞競合を駆動する
山本真寿・井垣達吏
(京都大学大学院生命科学研究科 高次生命科学専攻システム機能学分野)
email:井垣達吏
DOI: 10.7875/first.author.2017.019
The ligand Sas and its receptor PTP10D drive tumour-suppressive cell competition.
Masatoshi Yamamoto, Shizue Ohsawa, Kei Kunimasa, Tatsushi Igaki
Nature, 542, 246-250 (2017)
がんのほとんどは上皮細胞に由来しており,上皮細胞の頂底軸方向の極性の崩壊とがんの進展とは正の相関を示す.実際に,上皮組織に生じた極性崩壊細胞は高い増殖能をもつが,近年,その周囲を正常な細胞にかこまれると細胞競合の敗者となり組織から排除されることがわかってきた.この細胞競合において,敗者となる極性崩壊細胞はEiger/TNF-JNKシグナル伝達系を介した細胞死により組織から排除されることが明らかにされていたが,正常な細胞は隣接する極性崩壊細胞をどのように認識して排除するのか,その最上流の分子機構はまったく不明であった.筆者らは,この研究において,ショウジョウバエを用いた大規模な遺伝学的なスクリーニングにより,正常な細胞に発現する細胞表面のリガンドSasと極性崩壊細胞に発現するその受容体PTP10Dがこの細胞競合を駆動することを明らかにした.さらに,その分子機構として,正常な細胞と極性崩壊細胞との境界面においてSasおよびPTP10Dの局在がそれぞれの側面膜へと変化し,これによりトランスに活性化されたPTP10Dが極性崩壊細胞のEGF受容体-Rasシグナル伝達系を抑制することによりEiger/TNF-JNKシグナルを細胞増殖シグナルから細胞死シグナルへと変換し,極性崩壊細胞の排除を駆動することが見い出された.
多細胞生物が個体としての生命機能を維持するためには,構成する個々の細胞の増殖や死,分化といったふるまいを厳密かつ協調的に制御する必要がある.この協調的な細胞制御の破綻の典型例ががん化であり,少数の細胞が際限なく増殖することにより組織や個体に重篤な影響を及ぼす.一方,正常な組織にはこのような非協調的な細胞を積極的に排除するしくみが備わっており,たとえば,イヌの腎臓尿細管の上皮細胞に由来するMDCK細胞やゼブラフィッシュの胚において,Ras遺伝子やSrc遺伝子などのがん遺伝子により形質転換された細胞は周囲を正常な細胞にかこまれると上皮組織から排除される1,2).つまり,正常な上皮組織には細胞間の相互作用を介したがんの抑制機構が内在すると考えられる.この組織に内在的ながんの抑制機構は細胞競合とよばれる現象により駆動されると考えられている.細胞競合とは,組織を構成する細胞のあいだで相対的に適応度の高い細胞が適応度の低い細胞を駆逐する現象であり,種々のがん遺伝子およびがん抑制遺伝子の変異によりひき起こされることが示されている3).ここ10年ほどの研究により,細胞競合の敗者となった細胞が組織から排除される分子機構については理解が進んできたが,勝者が敗者を認識する分子機構についてはまったくわかっていない.
筆者らは,この研究において,ショウジョウバエの上皮細胞をモデルとして用い,細胞競合の勝者は敗者をどのように認識し排除するのか,その分子機構を遺伝学的に解析した.
ヒトのがんのほとんどは上皮細胞に由来する.がんの進展と上皮細胞の頂底軸方向の極性の崩壊とは正に相関しており4),また,さまざまな実験系において極性の崩壊した上皮細胞は過剰な増殖能をもつことが示されている5).ショウジョウバエの上皮細胞においても同様に,進化的に保存された頂底軸方向の極性遺伝子であるscrib遺伝子やdlg遺伝子に変異が生じるとそれら極性崩壊細胞は過剰に増殖して腫瘍を形成する6).これら極性崩壊細胞は正常な上皮細胞に周囲をかこまれると,その高い増殖能を発揮することなく組織から積極的に排除される6,7).この現象は,細胞競合を介した上皮細胞に内在性のがんの抑制機構と考えられている.すなわち,正常な細胞と極性崩壊細胞とが同一の組織に共存すると,正常な細胞が細胞競合の勝者となり敗者となる極性崩壊細胞を組織から排除する.これまで,筆者らを含む複数の研究グループにより,極性崩壊細胞はショウジョウバエにおいてはEiger/TNF-JNKシグナルに依存的な細胞死により組織から排除されることが明らかにされている6-8)(文献8) は新着論文レビュー でも掲載).しかし,周囲の正常な細胞がいかにして極性崩壊細胞を認識して排除するのか,その分子機構はまったく不明であった.
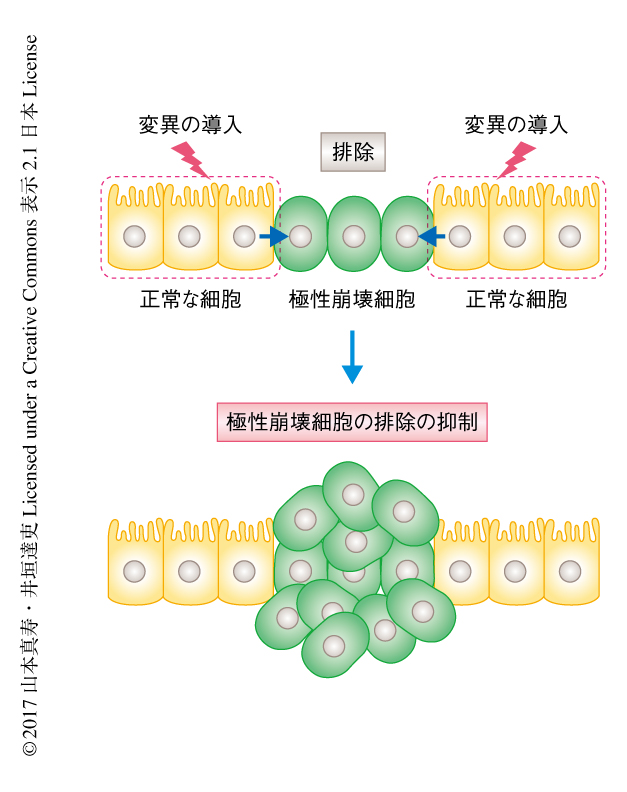
そこで,ショウジョウバエを用いた大規模な遺伝学的なスクリーニングにより,極性崩壊細胞の認識および排除に必要となる正常な細胞の側のタンパク質の同定を試みた.具体的には,ショウジョウバエの上皮組織である複眼成虫原基に正常な細胞のクローンと極性崩壊細胞のクローンをモザイク状に誘導し,変異原化合物エチルメタンスルホン酸により正常な細胞のクローンにのみランダムな変異を導入した.約7500の変異系統を樹立してスクリーニングした結果,極性崩壊細胞を排除できなくなる変異系統を8つ取得することに成功した(図1).遺伝学的な手法およびDNA塩基配列の解析により,これらの変異系統のうち4系統の原因遺伝子はいずれもsas遺伝子であることがわかった.sas遺伝子は細胞の表面に存在する1回膜貫通型のリガンド様のタンパク質をコードしており,これまで,ショウジョウバエにおいて神経軸索の誘導に関与することが報告されていたが9),上皮細胞における役割は不明であった.
正常な細胞が隣接する極性崩壊細胞を排除するのに細胞表面のリガンドSasが必要であることがわかった.では,Sasはどのように隣接する極性崩壊細胞の排除に寄与するのだろうか? 細胞におけるSasの局在を解析した結果,正常な上皮組織においては細胞の頂端膜に局在するのに対し,正常な細胞と極性崩壊細胞との境界面においては側面膜へとその局在が変化することがわかった.さらなる解析により,このSasの側面膜への局在の変化が起こる原因は,極性崩壊細胞と接する正常な細胞の頂端膜が拡大して細胞の境界面の側面膜へと落ち込むためと考えられた.Sasを欠失させた極性崩壊細胞のクローンにおいても正常な細胞と極性崩壊細胞との境界面へのSasの局在の変化がみられたことから,この境界面へのSasの集積は極性崩壊細胞に隣接する正常な細胞において起こることがわかった.
Sasはその細胞外領域にFN3ドメインおよびVWCドメインという2つのタンパク質間相互作用ドメインをもつ.このことに着目して,極性崩壊細胞の側でSasの受容体として機能するタンパク質を探索した.ショウジョウバエのゲノムのデータベースから,FN3ドメインあるいはVWCドメインをもつ膜タンパク質をコードする遺伝子を候補として絞り込んだ.そして,極性崩壊細胞において32個の候補遺伝子をRNAi法を用いてノックダウンし,極性崩壊細胞の排除が抑制されるものを探索した.その結果,極性崩壊細胞において受容体型チロシンホスファターゼをコードするPtp10d遺伝子をノックダウンすると極性崩壊細胞の排除が顕著に抑制されることがわかった.Ptp10d遺伝子をノックダウンした極性崩壊細胞は排除をまぬがれるだけでなく,大過剰に増殖して組織に腫瘍を形成した.
これまでに,ニューロンに存在するPTP10Dがグリア細胞に存在するSasとトランスに結合して神経軸索の誘導を制御することが報告されていた9).このことから,上皮組織においてもSasとPTP10Dがリガンドと受容体として相互作用し,細胞競合における細胞の認識および排除に寄与する可能性が強く示唆された.実際に,PTP10DもSasと同様に正常な細胞と極性崩壊細胞との境界面の側面膜へと局在が変化した.極性崩壊細胞においてPTP10Dをノックダウンすると境界面に局在するPTP10Dは顕著に減少したが,周囲の正常な細胞においてPTP10Dをノックダウンしても境界面に局在するPTP10Dは変化しなかった.これらのことから,境界面に局在するPTP10Dは極性崩壊細胞の側に由来すると考えられた.
ここまでの結果から,正常な細胞のSasと極性崩壊細胞のPTP10Dとの境界面におけるトランスな相互作用が極性崩壊細胞の排除を担うと考えられた.
SasとPTP10Dとのトランスな活性化を介して極性崩壊細胞はいかにして排除されるのか,そのシグナル伝達経路の同定を試みた.以前の研究により,極性崩壊細胞におけるEiger/TNF-JNKシグナル伝達経路の活性化がその排除に必須であることがわかっていた6-8).そこで,Sas-PTP10DシグナルとJNKシグナルの活性化との関係について調べた.その結果,PTP10Dのノックダウンにより排除の抑制された極性崩壊細胞においてもJNKシグナルは強く活性化されたままであることがわかった.つまり,Sas-PTP10DシグナルはJNKシグナルに依存的な細胞死を直接的に制御して極性崩壊細胞の排除に寄与するのではないことがわかった.
以前の研究により,PTP10DはEGF受容体の細胞内ドメインを直接に脱リン酸化することによりEGF受容体-Rasシグナルを抑制することが示されていた10).実際に,PTP10Dのノックダウンにより排除の抑制された極性崩壊細胞においてはEGF受容体-Rasシグナルの顕著な上昇が観察された.すなわち,正常な細胞と極性崩壊細胞との境界面においてSasにより活性化されたPTP10Dが,極性崩壊細胞においてEGF受容体を抑制している可能性が考えられた.筆者らは,以前の研究において,正常な細胞においては細胞死の誘導にはたらくJNKシグナルが,Rasシグナルの活性化により繊維状アクチンの集積を介してがんの抑制にはたらくHippoシグナル伝達経路を不活性化することにより,過剰な増殖および腫瘍の形成をひき起こすシグナルに変換されることを見い出していた11,12)(文献12) は新着論文レビュー でも掲載).つまり,Sas-PTP10Dシグナルがはたらかず排除の抑制された極性崩壊細胞においては,抑制がはずれて活性化したEGF受容体-RasシグナルがJNKシグナルを細胞死シグナルから細胞増殖シグナルへと変換すると予想された.
このことを裏づけるように,Sas-PTP10Dシグナルがはたらかず排除の抑制された極性崩壊細胞においては,細胞における繊維状アクチンの異常な集積およびHippoシグナル伝達経路の不活性化が観察された.また,EGF受容体のノックダウンやRasドミナントネガティブ変異体によるEGF受容体-Rasシグナルの抑制により,Sas-PTP10Dシグナルの不全による極性崩壊細胞の排除の抑制および過剰な増殖は顕著に抑制された.さらに,Hippoシグナル伝達経路を構成するキナーゼWtsによりHippoシグナル伝達経路を活性化させることによっても,Sas-PTP10Dシグナルの不全による極性崩壊細胞の排除の抑制および過剰な増殖は抑制された.
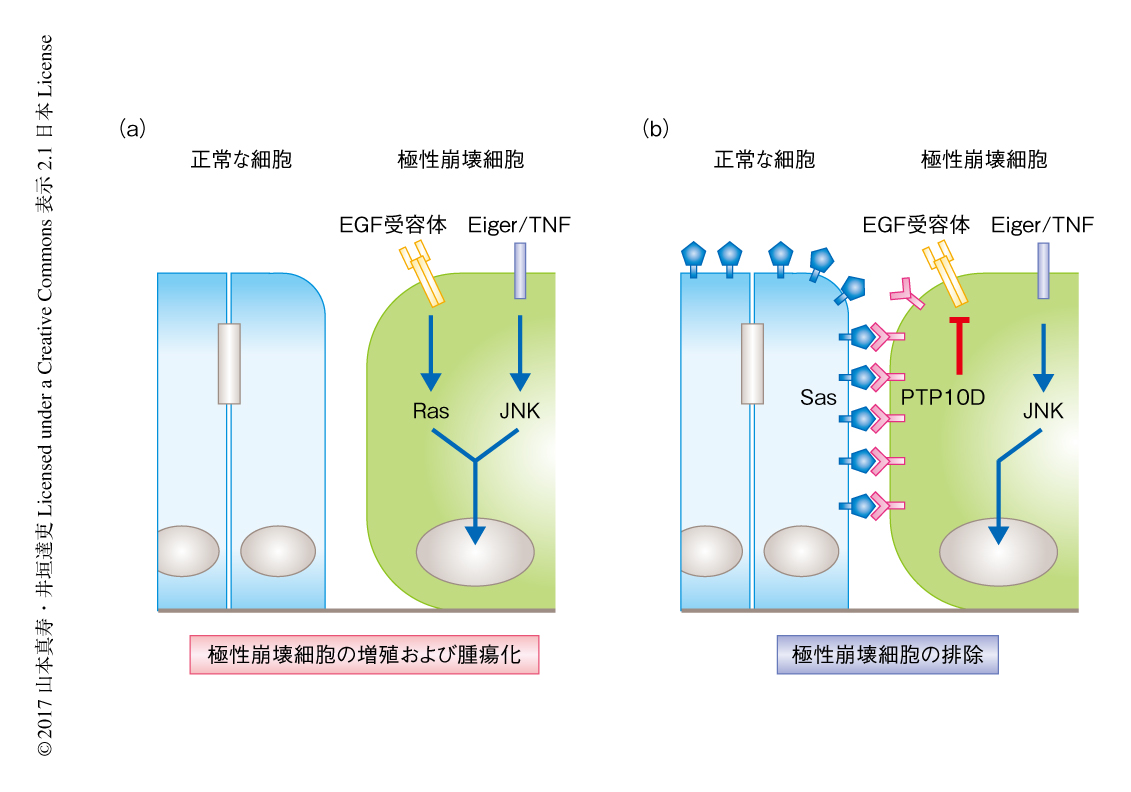
以上の結果から,以下のようなモデルが考えられた.すなわち,上皮細胞における極性の崩壊はEiger/TNF-JNKシグナルおよびEGF受容体-Rasシグナルの活性化をひき起こし,細胞に高いがん原性を付与する.しかし,そのような極性崩壊細胞が出現すると隣接する正常な細胞においてSasの局在が境界面へと変化し,それに応じるように極性崩壊細胞においてもPTP10Dの局在が境界面へと変化するため,この境界面においてSas-PTP10Dシグナルのトランスな活性化が起こる.これにより,極性崩壊細胞においてはEGF受容体-Rasシグナルが抑制され,JNKシグナルが細胞死の誘導にはたらくことにより,極性崩壊細胞は組織から排除されると考えられる(図2).
この研究により,上皮組織に出現したがん原性の極性崩壊細胞を排除する,がんの抑制にはたらく細胞競合を担う細胞表面のリガンドSasと受容体PTP10Dが明らかにされた.Sas-PTP10Dシグナルは頂底軸極性の崩壊した細胞と正常な極性をもつ細胞とが隣接することによりはじめて活性化すると考えられる.すなわち,極性の崩壊したがん原性をもつ細胞が正常な組織に生じると,Sas-PTP10Dシグナルが局所的にはたらいて極性崩壊細胞のEGF受容体-Rasシグナルを抑制し,JNKシグナルを細胞死シグナルへと変換することにより極性崩壊細胞を排除する.このように,Sas-PTP10Dシグナルは上皮組織のがん化をふせぐ安全装置として機能すると考えられる.細胞競合による極性崩壊細胞の排除は哺乳類の培養細胞系においても示されており13),PTP10Dの哺乳類におけるホモログをコードするPTPRJ遺伝子もがん抑制遺伝子として機能することが報告されている14).今回,見い出されたSas-PTP10Dシグナルに相当するしくみが哺乳類においても保存されていれば,これに着目した新たながんの治療および予防の戦略への応用が期待できる.
研究テーマ:がんの抑制にはたらく細胞競合を駆動する分子機構.
関心事:がん細胞と正常な細胞との相互作用,細胞のがん化における生体膜の構造の変化の意義.
井垣 達吏(Tatsushi Igaki)
京都大学大学院生命科学研究科 教授.
研究室URL:http://www.lif.kyoto-u.ac.jp/genetics/
© 2017 山本真寿・井垣達吏 Licensed under CC 表示 2.1 日本
(京都大学大学院生命科学研究科 高次生命科学専攻システム機能学分野)
email:井垣達吏
DOI: 10.7875/first.author.2017.019
The ligand Sas and its receptor PTP10D drive tumour-suppressive cell competition.
Masatoshi Yamamoto, Shizue Ohsawa, Kei Kunimasa, Tatsushi Igaki
Nature, 542, 246-250 (2017)
要 約
がんのほとんどは上皮細胞に由来しており,上皮細胞の頂底軸方向の極性の崩壊とがんの進展とは正の相関を示す.実際に,上皮組織に生じた極性崩壊細胞は高い増殖能をもつが,近年,その周囲を正常な細胞にかこまれると細胞競合の敗者となり組織から排除されることがわかってきた.この細胞競合において,敗者となる極性崩壊細胞はEiger/TNF-JNKシグナル伝達系を介した細胞死により組織から排除されることが明らかにされていたが,正常な細胞は隣接する極性崩壊細胞をどのように認識して排除するのか,その最上流の分子機構はまったく不明であった.筆者らは,この研究において,ショウジョウバエを用いた大規模な遺伝学的なスクリーニングにより,正常な細胞に発現する細胞表面のリガンドSasと極性崩壊細胞に発現するその受容体PTP10Dがこの細胞競合を駆動することを明らかにした.さらに,その分子機構として,正常な細胞と極性崩壊細胞との境界面においてSasおよびPTP10Dの局在がそれぞれの側面膜へと変化し,これによりトランスに活性化されたPTP10Dが極性崩壊細胞のEGF受容体-Rasシグナル伝達系を抑制することによりEiger/TNF-JNKシグナルを細胞増殖シグナルから細胞死シグナルへと変換し,極性崩壊細胞の排除を駆動することが見い出された.
はじめに
多細胞生物が個体としての生命機能を維持するためには,構成する個々の細胞の増殖や死,分化といったふるまいを厳密かつ協調的に制御する必要がある.この協調的な細胞制御の破綻の典型例ががん化であり,少数の細胞が際限なく増殖することにより組織や個体に重篤な影響を及ぼす.一方,正常な組織にはこのような非協調的な細胞を積極的に排除するしくみが備わっており,たとえば,イヌの腎臓尿細管の上皮細胞に由来するMDCK細胞やゼブラフィッシュの胚において,Ras遺伝子やSrc遺伝子などのがん遺伝子により形質転換された細胞は周囲を正常な細胞にかこまれると上皮組織から排除される1,2).つまり,正常な上皮組織には細胞間の相互作用を介したがんの抑制機構が内在すると考えられる.この組織に内在的ながんの抑制機構は細胞競合とよばれる現象により駆動されると考えられている.細胞競合とは,組織を構成する細胞のあいだで相対的に適応度の高い細胞が適応度の低い細胞を駆逐する現象であり,種々のがん遺伝子およびがん抑制遺伝子の変異によりひき起こされることが示されている3).ここ10年ほどの研究により,細胞競合の敗者となった細胞が組織から排除される分子機構については理解が進んできたが,勝者が敗者を認識する分子機構についてはまったくわかっていない.
筆者らは,この研究において,ショウジョウバエの上皮細胞をモデルとして用い,細胞競合の勝者は敗者をどのように認識し排除するのか,その分子機構を遺伝学的に解析した.
1.細胞表面のリガンドSasは隣接する極性崩壊細胞の排除に必要である
ヒトのがんのほとんどは上皮細胞に由来する.がんの進展と上皮細胞の頂底軸方向の極性の崩壊とは正に相関しており4),また,さまざまな実験系において極性の崩壊した上皮細胞は過剰な増殖能をもつことが示されている5).ショウジョウバエの上皮細胞においても同様に,進化的に保存された頂底軸方向の極性遺伝子であるscrib遺伝子やdlg遺伝子に変異が生じるとそれら極性崩壊細胞は過剰に増殖して腫瘍を形成する6).これら極性崩壊細胞は正常な上皮細胞に周囲をかこまれると,その高い増殖能を発揮することなく組織から積極的に排除される6,7).この現象は,細胞競合を介した上皮細胞に内在性のがんの抑制機構と考えられている.すなわち,正常な細胞と極性崩壊細胞とが同一の組織に共存すると,正常な細胞が細胞競合の勝者となり敗者となる極性崩壊細胞を組織から排除する.これまで,筆者らを含む複数の研究グループにより,極性崩壊細胞はショウジョウバエにおいてはEiger/TNF-JNKシグナルに依存的な細胞死により組織から排除されることが明らかにされている6-8)(文献8) は新着論文レビュー でも掲載).しかし,周囲の正常な細胞がいかにして極性崩壊細胞を認識して排除するのか,その分子機構はまったく不明であった.
そこで,ショウジョウバエを用いた大規模な遺伝学的なスクリーニングにより,極性崩壊細胞の認識および排除に必要となる正常な細胞の側のタンパク質の同定を試みた.具体的には,ショウジョウバエの上皮組織である複眼成虫原基に正常な細胞のクローンと極性崩壊細胞のクローンをモザイク状に誘導し,変異原化合物エチルメタンスルホン酸により正常な細胞のクローンにのみランダムな変異を導入した.約7500の変異系統を樹立してスクリーニングした結果,極性崩壊細胞を排除できなくなる変異系統を8つ取得することに成功した(図1).遺伝学的な手法およびDNA塩基配列の解析により,これらの変異系統のうち4系統の原因遺伝子はいずれもsas遺伝子であることがわかった.sas遺伝子は細胞の表面に存在する1回膜貫通型のリガンド様のタンパク質をコードしており,これまで,ショウジョウバエにおいて神経軸索の誘導に関与することが報告されていたが9),上皮細胞における役割は不明であった.
2.正常な細胞においてSasの局在は極性崩壊細胞との境界面の側面膜に変化する
正常な細胞が隣接する極性崩壊細胞を排除するのに細胞表面のリガンドSasが必要であることがわかった.では,Sasはどのように隣接する極性崩壊細胞の排除に寄与するのだろうか? 細胞におけるSasの局在を解析した結果,正常な上皮組織においては細胞の頂端膜に局在するのに対し,正常な細胞と極性崩壊細胞との境界面においては側面膜へとその局在が変化することがわかった.さらなる解析により,このSasの側面膜への局在の変化が起こる原因は,極性崩壊細胞と接する正常な細胞の頂端膜が拡大して細胞の境界面の側面膜へと落ち込むためと考えられた.Sasを欠失させた極性崩壊細胞のクローンにおいても正常な細胞と極性崩壊細胞との境界面へのSasの局在の変化がみられたことから,この境界面へのSasの集積は極性崩壊細胞に隣接する正常な細胞において起こることがわかった.
3.極性崩壊細胞はSasの受容体であるPTP10Dを介して排除される
Sasはその細胞外領域にFN3ドメインおよびVWCドメインという2つのタンパク質間相互作用ドメインをもつ.このことに着目して,極性崩壊細胞の側でSasの受容体として機能するタンパク質を探索した.ショウジョウバエのゲノムのデータベースから,FN3ドメインあるいはVWCドメインをもつ膜タンパク質をコードする遺伝子を候補として絞り込んだ.そして,極性崩壊細胞において32個の候補遺伝子をRNAi法を用いてノックダウンし,極性崩壊細胞の排除が抑制されるものを探索した.その結果,極性崩壊細胞において受容体型チロシンホスファターゼをコードするPtp10d遺伝子をノックダウンすると極性崩壊細胞の排除が顕著に抑制されることがわかった.Ptp10d遺伝子をノックダウンした極性崩壊細胞は排除をまぬがれるだけでなく,大過剰に増殖して組織に腫瘍を形成した.
これまでに,ニューロンに存在するPTP10Dがグリア細胞に存在するSasとトランスに結合して神経軸索の誘導を制御することが報告されていた9).このことから,上皮組織においてもSasとPTP10Dがリガンドと受容体として相互作用し,細胞競合における細胞の認識および排除に寄与する可能性が強く示唆された.実際に,PTP10DもSasと同様に正常な細胞と極性崩壊細胞との境界面の側面膜へと局在が変化した.極性崩壊細胞においてPTP10Dをノックダウンすると境界面に局在するPTP10Dは顕著に減少したが,周囲の正常な細胞においてPTP10Dをノックダウンしても境界面に局在するPTP10Dは変化しなかった.これらのことから,境界面に局在するPTP10Dは極性崩壊細胞の側に由来すると考えられた.
ここまでの結果から,正常な細胞のSasと極性崩壊細胞のPTP10Dとの境界面におけるトランスな相互作用が極性崩壊細胞の排除を担うと考えられた.
4.Sas-PTP10Dシグナルは極性崩壊細胞におけるJNKシグナルの活性化には影響しない
SasとPTP10Dとのトランスな活性化を介して極性崩壊細胞はいかにして排除されるのか,そのシグナル伝達経路の同定を試みた.以前の研究により,極性崩壊細胞におけるEiger/TNF-JNKシグナル伝達経路の活性化がその排除に必須であることがわかっていた6-8).そこで,Sas-PTP10DシグナルとJNKシグナルの活性化との関係について調べた.その結果,PTP10Dのノックダウンにより排除の抑制された極性崩壊細胞においてもJNKシグナルは強く活性化されたままであることがわかった.つまり,Sas-PTP10DシグナルはJNKシグナルに依存的な細胞死を直接的に制御して極性崩壊細胞の排除に寄与するのではないことがわかった.
5.Sas-PTP10Dシグナルは極性崩壊細胞のEGF受容体-Rasシグナルを抑制することによりJNKシグナルを細胞死シグナルに変換する
以前の研究により,PTP10DはEGF受容体の細胞内ドメインを直接に脱リン酸化することによりEGF受容体-Rasシグナルを抑制することが示されていた10).実際に,PTP10Dのノックダウンにより排除の抑制された極性崩壊細胞においてはEGF受容体-Rasシグナルの顕著な上昇が観察された.すなわち,正常な細胞と極性崩壊細胞との境界面においてSasにより活性化されたPTP10Dが,極性崩壊細胞においてEGF受容体を抑制している可能性が考えられた.筆者らは,以前の研究において,正常な細胞においては細胞死の誘導にはたらくJNKシグナルが,Rasシグナルの活性化により繊維状アクチンの集積を介してがんの抑制にはたらくHippoシグナル伝達経路を不活性化することにより,過剰な増殖および腫瘍の形成をひき起こすシグナルに変換されることを見い出していた11,12)(文献12) は新着論文レビュー でも掲載).つまり,Sas-PTP10Dシグナルがはたらかず排除の抑制された極性崩壊細胞においては,抑制がはずれて活性化したEGF受容体-RasシグナルがJNKシグナルを細胞死シグナルから細胞増殖シグナルへと変換すると予想された.
このことを裏づけるように,Sas-PTP10Dシグナルがはたらかず排除の抑制された極性崩壊細胞においては,細胞における繊維状アクチンの異常な集積およびHippoシグナル伝達経路の不活性化が観察された.また,EGF受容体のノックダウンやRasドミナントネガティブ変異体によるEGF受容体-Rasシグナルの抑制により,Sas-PTP10Dシグナルの不全による極性崩壊細胞の排除の抑制および過剰な増殖は顕著に抑制された.さらに,Hippoシグナル伝達経路を構成するキナーゼWtsによりHippoシグナル伝達経路を活性化させることによっても,Sas-PTP10Dシグナルの不全による極性崩壊細胞の排除の抑制および過剰な増殖は抑制された.
以上の結果から,以下のようなモデルが考えられた.すなわち,上皮細胞における極性の崩壊はEiger/TNF-JNKシグナルおよびEGF受容体-Rasシグナルの活性化をひき起こし,細胞に高いがん原性を付与する.しかし,そのような極性崩壊細胞が出現すると隣接する正常な細胞においてSasの局在が境界面へと変化し,それに応じるように極性崩壊細胞においてもPTP10Dの局在が境界面へと変化するため,この境界面においてSas-PTP10Dシグナルのトランスな活性化が起こる.これにより,極性崩壊細胞においてはEGF受容体-Rasシグナルが抑制され,JNKシグナルが細胞死の誘導にはたらくことにより,極性崩壊細胞は組織から排除されると考えられる(図2).
おわりに
この研究により,上皮組織に出現したがん原性の極性崩壊細胞を排除する,がんの抑制にはたらく細胞競合を担う細胞表面のリガンドSasと受容体PTP10Dが明らかにされた.Sas-PTP10Dシグナルは頂底軸極性の崩壊した細胞と正常な極性をもつ細胞とが隣接することによりはじめて活性化すると考えられる.すなわち,極性の崩壊したがん原性をもつ細胞が正常な組織に生じると,Sas-PTP10Dシグナルが局所的にはたらいて極性崩壊細胞のEGF受容体-Rasシグナルを抑制し,JNKシグナルを細胞死シグナルへと変換することにより極性崩壊細胞を排除する.このように,Sas-PTP10Dシグナルは上皮組織のがん化をふせぐ安全装置として機能すると考えられる.細胞競合による極性崩壊細胞の排除は哺乳類の培養細胞系においても示されており13),PTP10Dの哺乳類におけるホモログをコードするPTPRJ遺伝子もがん抑制遺伝子として機能することが報告されている14).今回,見い出されたSas-PTP10Dシグナルに相当するしくみが哺乳類においても保存されていれば,これに着目した新たながんの治療および予防の戦略への応用が期待できる.
文 献
- Hogan, C., Dupre-Crochet, S., Norman, M. et al.: Characterization of the interface between normal and transformed epithelial cells. Nat. Cell Biol., 11, 460-467 (2009)[PubMed]
- Kajita, M., Hogan, C., Harris, A. R. et al.: Interaction with surrounding normal epithelial cells influences signalling pathways and behaviour of Src-transformed cells. J. Cell Sci., 123, 171-180 (2010)[PubMed]
- Amoyel, M. & Bach, E. A.: Cell competition: how to eliminate your neighbours. Development, 141, 988-1000 (2014)[PubMed]
- Martin-Belmonte, F. & Perez-Moreno, M.: Epithelial cell polarity, stem cells and cancer. Nat. Rev. Cancer, 12, 23-38 (2012)[PubMed]
- Huang, L. & Muthuswamy, S. K.: Polarity protein alterations in carcinoma: a focus on emerging roles for polarity regulators. Curr. Opin. Genet. Dev., 20, 41-50 (2010)[PubMed]
- Brumby, A. M. & Richardson, H. E.: scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J., 22, 5769-5779 (2003)[PubMed]
- Igaki, T., Pastor-Pareja, J. C., Aonuma, H. et al.: Intrinsic tumor suppression and epithelial maintenance by endocytic activation of Eiger/TNF signaling in Drosophila. Dev. Cell, 16, 458-465 (2009)[PubMed]
- Ohsawa, S., Sugimura, K., Takino, K. et al.: Elimination of oncogenic neighbors by JNK-mediated engulfment in Drosophila. Dev. Cell, 20, 315-328 (2011)[PubMed] [新着論文レビュー]
- Lee, H. K., Cording, A., Vielmetter, J. et al.: Interactions between a receptor tyrosine phosphatase and a cell surface ligand regulate axon guidance and glial-neuronal communication. Neuron, 78, 813-826 (2013)[PubMed]
- Jeon, M. & Zinn, K.: Receptor tyrosine phosphatases control tracheal tube geometries through negative regulation of Egfr signaling. Development, 136, 3121-3129 (2009)[PubMed]
- Enomoto, M., Kizawa, D., Ohsawa, S. et al.: JNK signaling is converted from anti- to pro-tumor pathway by Ras-mediated switch of Warts activity. Dev. Biol., 403, 162-171 (2015)[PubMed]
- Ohsawa, S., Sato, Y., Enomoto, M. et al.: Mitochondrial defect drives non-autonomous tumour progression through Hippo signalling in Drosophila. Nature, 490, 547-551 (2012)[PubMed] [新着論文レビュー]
- Norman, M., Wisniewska, K. A., Lawrenson, K. et al.: Loss of Scribble causes cell competition in mammalian cells. J. Cell Sci., 125, 59-66 (2012)[PubMed]
- Hendriks, W. J. A. J. & Pulido, R.: Protein tyrosine phosphatase variants in human hereditary disorders and disease susceptibilities. Biochim. Biophys. Acta, 1832, 1673-1696 (2013)[PubMed]
活用したデータベースにかかわるキーワードと統合TVへのリンク
著者プロフィール
研究テーマ:がんの抑制にはたらく細胞競合を駆動する分子機構.
関心事:がん細胞と正常な細胞との相互作用,細胞のがん化における生体膜の構造の変化の意義.
井垣 達吏(Tatsushi Igaki)
京都大学大学院生命科学研究科 教授.
研究室URL:http://www.lif.kyoto-u.ac.jp/genetics/
© 2017 山本真寿・井垣達吏 Licensed under CC 表示 2.1 日本
