肝臓に由来する分泌タンパク質selenoprotein Pはインスリン抵抗性の原因となる
御簾博文・篁 俊成
(金沢大学医薬保健研究域医学類 恒常性制御学)
email:御簾博文,篁 俊成
DOI: 10.7875/first.author.2010.056
A liver-derived secretory protein, selenoprotein P, causes insulin resistance.
Hirofumi Misu, Toshinari Takamura, Hiroaki Takayama, Hiroto Hayashi, Naoto Matsuzawa-Nagata, Seiichiro Kurita, Kazuhide Ishikura, Hitoshi Ando, Yumie Takeshita, Tsuguhito Ota, Masaru Sakurai, Tatsuya Yamashita, Eishiro Mizukoshi, Taro Yamashita, Masao Honda, Ken-ichi Miyamoto, Tetsuya Kubota, Naoto Kubota, Takashi Kadowaki, Han-Jong Kim, In-kyu Lee, Yasuhiko Minokoshi, Yoshiro Saito, Kazuhiko Takahashi, Yoshihiro Yamada, Nobuyuki Takakura, Shuichi Kaneko
Cell Metabolism, 12, 483-495 (2010)
肝臓はヘパトカインとよばれる分泌タンパク質の産生を介して全身のインスリン感受性を修飾することで糖の恒常性を制御している可能性がある.筆者らは,今回,肝臓に由来する分泌タンパク質のひとつであるselenoprotein Pがインスリン抵抗性の原因となることを明らかにした.まず,ヒト肝臓でのselenoprotein Pの遺伝子発現量がインスリン抵抗性と相関することを見い出した.精製selenoprotein Pによる処理は肝細胞と筋細胞の両方でインスリンシグナルを障害し糖代謝の異常を誘導した.逆に,selenoprotein PのノックアウトならびにRNAiによる抑制ノックダウンはマウスにおいて全身のインスリン感受性と耐糖能を改善した.selenoprotein Pの代謝作用の少なくとも一部はAMPキナーゼの不活性化によるものであった.今回の結果は,selenoprotein Pの糖代謝およびインスリン感受性の制御におけるこれまで知られていなかった役割を明らかにし,selenoprotein Pが2型糖尿病を代表とするインスリン抵抗性関連疾患の治療標的になることを示唆した.
インスリン抵抗性は2型糖尿病患者およびメタボリックシンドローム患者の基盤にある病態であり1),心血管系疾患や非アルコール性肝疾患の発症にも関連している.インスリン抵抗性状態ではインスリン作用の障害が肝臓での糖産生の亢進と末梢組織での糖取り込みの低下を誘導し,その結果として高血糖が生じる.インスリン抵抗性の分子機構は完全には理解されていないが,これまで古典的に内分泌臓器と考えられていた臓器とは異なる臓器から分泌される液性因子が,その発症に影響することが知られるようになってきた.たとえば,最近の肥満研究は,脂肪組織はアディポサイトカインとよばれる種々のタンパク質を分泌すること,このアディポサイトカインの産生異常がインスリン抵抗性の発症に寄与することを明らかにしている2).
selenoprotein Pはおもに肝臓で産生される分泌タンパク質である3).10残基のセレノシステイン残基を含んでおり,微量元素であるセレンの輸送タンパク質として機能する4).しかし,その糖代謝ならびにインスリン感受性の制御における役割はこれまで確立されていなかった.さらに,selenoprotein Pノックアウトマウスの研究からその欠損が脳神経障害や不妊と関連することは報告されていたが5,6),ヒト疾患における臨床的な重要性はよくわかっていなかった.
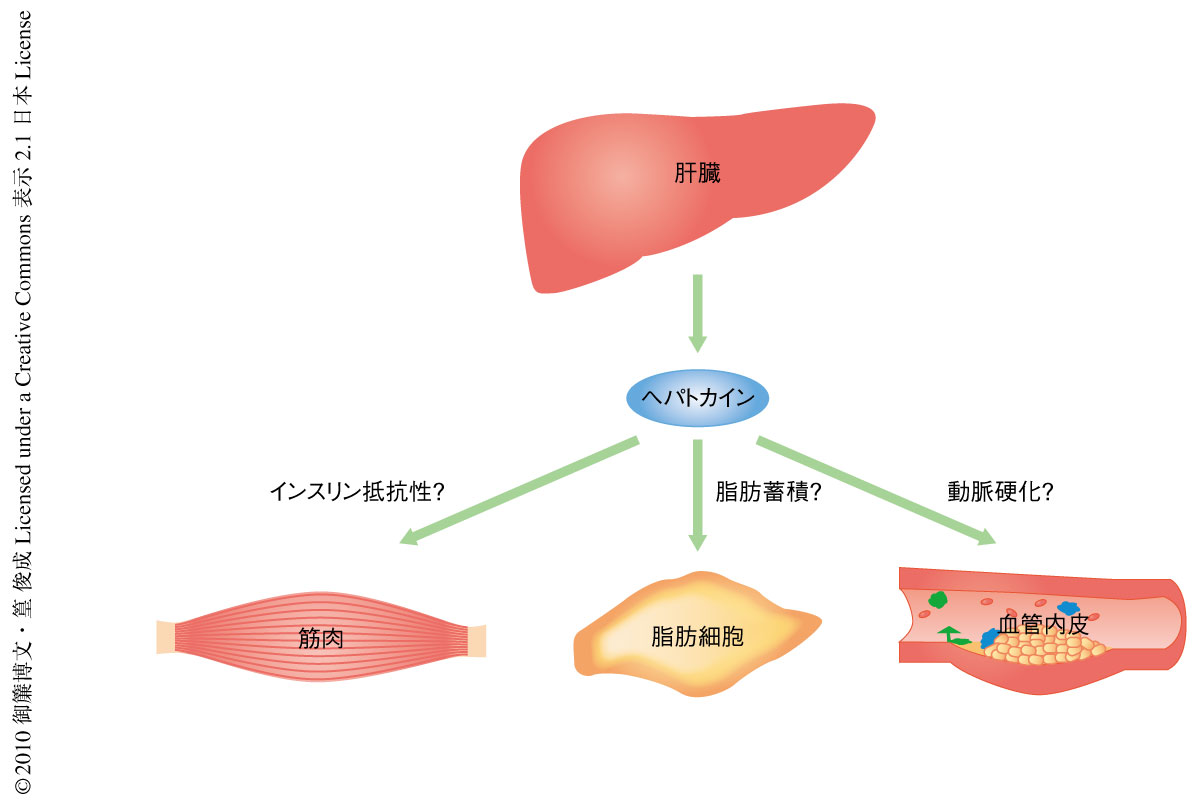
肝臓は糖の恒常性の制御において中心的な役割を担うとともに,多種多様な分泌タンパク質の産生臓器でもある.筆者らは,過去に,2型糖尿病患者の肝臓は分泌タンパク質をコードする膨大な種類の遺伝子を発現していることを報告した7).さらに,2型糖尿病患者の肝臓では血管新生因子,線維化促進因子,酸化ストレス関連因子の発現が健常者とは異なっており8),この変化は2型糖尿病患者の病態形成に貢献していることが推察された.これらの所見を根拠として,脂肪組織と同様に,肝臓が分泌タンパク質の産生を介して2型糖尿病やインスリン抵抗性の発症に寄与しているとの仮説をたてた.このような肝臓に由来する分泌タンパク質をヘパトカインと命名し,インスリン抵抗性関連ヘパトカインの探索を行った(図1).
インスリン抵抗性に関連した肝臓に由来する分泌タンパク質を同定する目的で,ヒト肝生検を行い肝臓に発現する遺伝子を2つの異なった方法を用いて包括的に解析した.まず,2型糖尿病患者5名と健常者5名に対して肝生検を行い,SAGE(serial analysis of gene expression)法を用いて発現遺伝子を包括的に解析した.その結果,分泌タンパク質をコードする遺伝子のうち健常者と比較して2型糖尿病患者で1.5倍以上も発現が変動している117種類の遺伝子を同定した.つぎに,2型糖尿病患者10名と健常者7名に対して超音波ガイドのもとで経皮的な肝生検を行い,DNAチップ解析によって肝臓における遺伝子発現量がインスリン抵抗性と相関する遺伝子を同定した.その結果,SAGE法による解析ではselenoprotein Pの肝臓における遺伝子発現量が2型糖尿病状態では約8倍に上昇していること,DNAチップ解析ではselenoprotein Pの肝臓における遺伝子発現がインスリン感受性と負に相関していることを見い出した.さらに,selenoprotein Pの肝臓における遺伝子発現は2型糖尿病患者の負荷後血糖値あるいは空腹血糖値とも正に相関する傾向を認めた.
インスリン抵抗性の発症におけるselenoprotein Pの役割を明らかにするため,ヒト血液中のselenoprotein Pの濃度をELISA法により測定した.肝臓でのselenoprotein Pの遺伝子発現の上昇と一致して,血中selenoprotein P濃度は空腹血糖値とHbA1c値の両方と正に相関していた.HbA1cは過去2~3か月の血糖調節の状態を反映する臨床マーカーである.くわえて,2型糖尿病患者の血中selenoprotein P濃度は健常者と比較して上昇していた.ヒト由来のデータと一致して,2型糖尿病モデル動物であるOLETFラットおよびKKAyマウスにおいても肝臓でのselenoprotein Pの遺伝子発現量と血中濃度は上昇していた.
2型糖尿病におけるselenoprotein Pの上昇に寄与する病態を明らかにするため,いくつかの栄養素を肝細胞に処理してselenoprotein Pの遺伝子発現におよぼす影響を検討した.その結果,グルコースおよびパルミチン酸の添加が遺伝子発現を上昇させること,一方,インスリン処理が用量依存的および時間依存的に遺伝子発現を減弱することを見い出した.マウス初代培養肝細胞においてもselenoprotein Pの濃度について同様の結果が認められた.肝細胞におけるインスリンによる負の制御と一致して,空腹状態のマウスの肝臓ではselenoprotein Pの遺伝子発現の上昇が認められた.
selenoprotein Pの遺伝子過剰発現系の作製は技術的に困難をともなうため,クロマトグラフィー法でヒト血液からselenoprotein Pを精製して細胞に対する投与実験を行った.マウス初代肝細胞において精製selenoprotein P処理はインスリン刺激によるインスリン受容体およびその下流のAktのリン酸化を有意に減弱させた.さらに,同様の結果はC2C12筋細胞でも観察された.つぎに,selenoprotein Pが細胞内の糖代謝に異常を起こしうるかどうかを検討した.H4IIEC肝細胞においてselenoprotein Pは糖新生の律速酵素であるPEPCK1およびG6paseの遺伝子発現を上昇させ,その結果,インスリン存在下における肝細胞からの糖放出を30%も増加させた.くわえて,selenoprotein P処理はC2C12筋細胞においてインスリン刺激性の糖取り込みを有意に減弱させた.これらのin vitroでの実験結果は,生理的な濃度でのselenoprotein P処置が細胞のインスリンシグナル伝達を障害し糖代謝の異常を誘導することを明らかにした.
in vivoでのselenoprotein Pの糖代謝におよぼす影響を調べるため,C57BLマウスに精製ヒトselenoprotein Pを実験の12時間前と2時間前の2回,1 mg/kg体重を腹腔内投与した.マウスの血中ヒトselenoprotein P濃度は0.5~1.5μg/mlにいたった.この濃度は健常者と2型糖尿病患者との血中濃度の差分に相当する.糖負荷試験およびインスリン負荷試験は精製selenoprotein Pの投与はマウスに耐糖能の悪化とインスリン抵抗性を誘導することを明らかにした.インスリン誘導性のAktのリン酸化はselenoprotein P投与マウスの肝臓および骨格筋の両方で低下していた.また,精製selenoprotein Pの投与は内因性の糖産生を上昇させ末梢でのグルコース利用率を低下させることが示された.これらの結果は,in vivoにおいてselenoprotein Pが肝臓と骨格筋のインスリンシグナルを障害し耐糖能の異常を誘導することを明らかにした.
内因性のselenoprotein Pのノックダウンがインスリンシグナルを亢進するかどうかを決定するため,H4IIEC肝細胞にselenoprotein Pに特異的なsiRNAを導入した.このselenoprotein Pノックダウン細胞ではインスリン刺激によるAktのリン酸化が有意に増強した.同様に,ハイドロダイナミック法を用いて糖尿病マウスであるKKAyマウスの肝臓にselenoprotein Pに特異的なsiRNAを導入したところ,肝臓および血中のselenoprotein P濃度は約30%も減少した.肝臓でのselenoprotein PのノックダウンはKKAyマウスの耐糖の異常とインスリン抵抗性の両方を改善した.
さらに,selenoprotein P濃度を長期間にわたり低下させることの効果を確認するためselenoprotein Pノックアウトマウスを使用した.selenoprotein Pノックアウトマウスはセレンが十分に含まれた食事で飼育すると生存が可能で正常な体重を呈した.肝臓での脂肪蓄積や脂肪細胞の外観に変化はなかった.しかし,血糖は不変であったが食後インスリン濃度が低値であった.糖負荷試験では耐糖能は良好で,インスリン負荷試験では負荷後血糖は有意に低値であった.インスリンシグナルはselenoprotein Pホモノックアウトマウスの肝臓と骨格筋の両方で増強していた.さらに,selenoprotein Pヘテロノックアウトマウスはインスリン感受性の増強している傾向を認めた.
selenoprotein Pの欠損が食事誘導性肥満によるインスリン抵抗性を減少させるかどうかを明らかにするため,selenoprotein Pノックアウトマウスを高脂肪高ショ糖食で飼育した.体重推移,肝中性脂肪含有量,脂肪組織重量には差を認めなかった.しかし,食事誘導性の脂肪細胞の肥大は軽度であった.空腹時血中インスリン濃度および遊離脂肪酸濃度は有意に低値であった.糖負荷試験およびインスリン負荷試験ではselenoprotein Pホモノックアウトマウスは肥満誘導食での飼育下においても耐糖能の異常とインスリン抵抗性から守られていた.
AMPキナーゼはさまざまなエネルギー関連タンパク質をリン酸化するセレンスレオニンキナーゼであり,インスリン感受性を促進するエネルギー制御タンパク質としてはたらく9).selenoprotein P処理はH4IIEC肝細胞とマウス肝臓の両方でAMPキナーゼαとその下流のACCのリン酸化を減弱させることが見い出された.脂肪酸のβ酸化とβ酸化関連遺伝子の発現もselenoprotein P処理で抑制された.selenoprotein Pを処理した肝細胞でAMPおよびATPの濃度は不変であった.機能阻害型AMPキナーゼを肝細胞に導入するとインスリン依存的なAktのリン酸化は減弱し,selenoprotein Pを処理してもこのリン酸化はさらなる低下を認めなかった.逆に,恒常活性型AMPキナーゼを過剰発現させるとselenoprotein Pのインスリン刺激性Aktリン酸化に対する抑制は消失した.これらの結果は,AMPキナーゼのリン酸化の減少がselenoprotein Pによるインスリン抵抗性の誘導の少なくとも一部を仲介することを示唆した.さらに,AMPキナーゼのリン酸化の上流タンパク質の発現を検討した.H4IIEC肝細胞においてAMPキナーゼのリン酸化の負の制御タンパク質であるプロテインホスファターゼ2Cの濃度はselenoprotein P処理によって用量依存的に増加した.
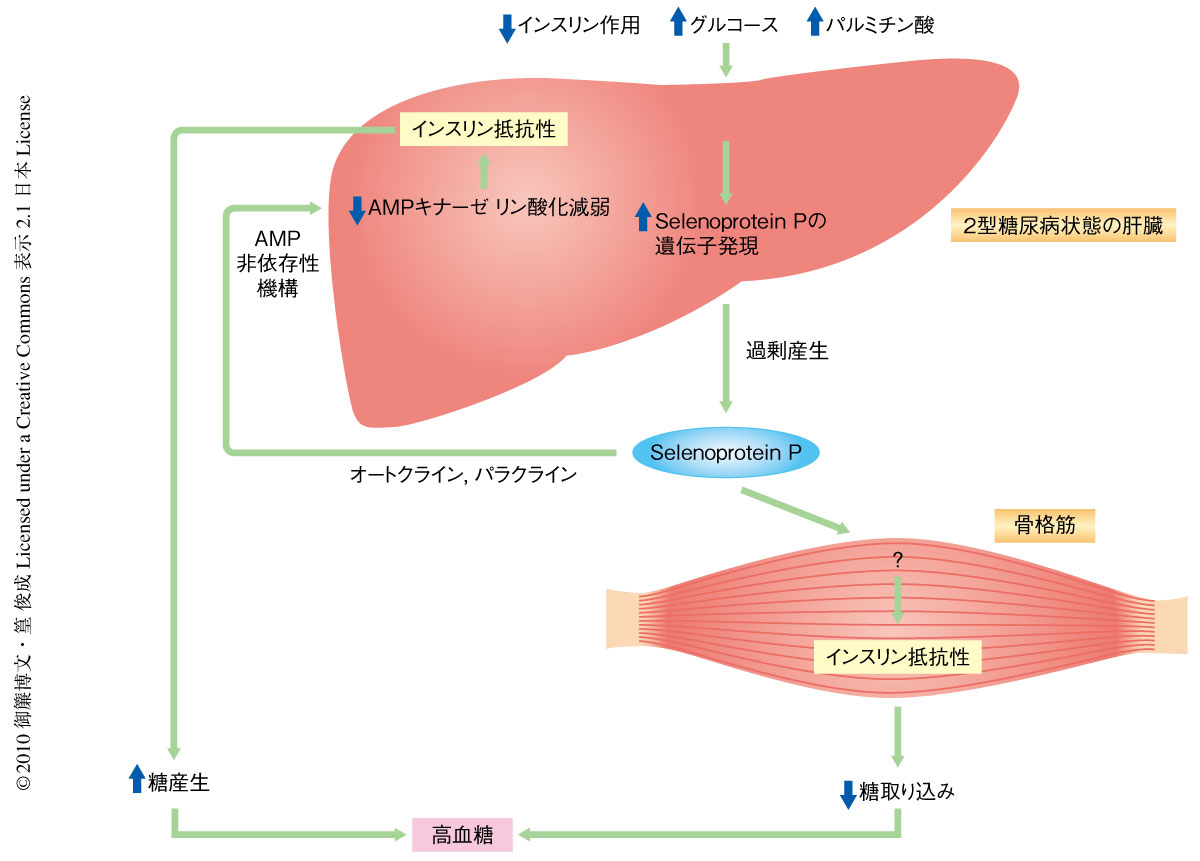
今回の研究は,肝臓でのselenoprotein Pの過剰産生が肝臓および骨格筋でのインスリン抵抗性の発症に寄与することを明らかにした(図2).肝臓はおもにグリコーゲンの貯蔵とグルコースの放出を介して糖の恒常性の維持に中心的な役割を担う.くわえて,肝臓は分泌タンパク質の大きな産生源である.ここから,筆者らは,肝臓は“ヘパトカイン”とよぶべき肝臓に由来する分泌タンパク質の産生を介して全身の糖の恒常性を維持するとの仮説をたてた.実際に,過去のいくつかの論文が肝臓に由来する分泌因子とインスリン抵抗性との関連を報告してきた.しかしながら,肝臓に由来しインスリン抵抗性や2型糖尿病の発症に寄与する液性因子の探索はいまだ十分ではないと推測した.なぜならば,筆者らの包括的な遺伝子発現解析では肝臓は分泌タンパク質をコードする膨大な種類の遺伝子を発現しており,その発現は糖尿病状態では変動していたからである7,8).こうして,肝臓における遺伝子発現量とインスリン抵抗性などの臨床パラメーターを比較することでインスリン抵抗性の原因となっている肝臓産生の分泌タンパク質の候補を選択した.この研究は,これまで知られていなかった脂肪組織と同様の肝臓の機能に光をあてた.すなわち,肝臓はホルモンの産生を介してインスリン抵抗性という病態の発症に寄与していたのである.
筆者らの結果は,インスリンが肝細胞におけるselenoprotein Pの発現を負に制御することを示した.この所見と一致して,selenoprotein Pの遺伝子発現はマウスにおいて空腹状態の肝臓で上昇することが示された.空腹状態に代表されるような低インスリン血症の状態では,selenoprotein Pの発現増加は末梢組織での糖取り込みの低下と肝での糖産生を増加させることで低血糖を予防している可能性がある.今回の研究は,肝臓が血液中のインスリン濃度を感知してselenoprotein Pの産生を変動させることで全身のインスリン感受性を制御しているという可能性を提示した.
インスリン標的器官におけるselenoprotein P受容体を同定することがselenoprotein Pによるインスリン抵抗性の誘導機構の解明には必要である.しかしながら,受容体の同定が技術的に困難であったため,今回の研究では糖代謝に作用しうる既知の経路とselenoprotein Pとの関連を探索した.その結果,肝細胞においてselenoprotein PがAMPキナーゼのリン酸化を減弱することを見い出した.さらに,AMPキナーゼリン酸化の負の制御タンパク質であるプロテインホスファターゼ2Cの濃度がselenoprotein Pによって上昇することを実証した.インスリン抵抗性に関連した代表的な炎症性サイトカインであるTNFαもプロテインホスファターゼ2Cの発現増加を介してAMPキナーゼを脱リン酸化することが報告されている10).今後,selenoprotein Pとselenoprotein P受容体との相互作用をさらに検討することで,selenoprotein Pによるプロテインホスファターゼ2Cの上方制御とAMPキナーゼのリン酸化の減少に関するさらなる洞察が得られるものと思われる.
おわりに,今回の研究はインスリン抵抗性および高血糖を誘導する肝臓由来の分泌タンパク質としてselenoprotein Pを同定した.この結果は,ヘパトカインであるselenoprotein Pが2型糖尿病を含むインスリン抵抗性関連疾患の治療標的となりうることを示唆した.
略歴:2007年 金沢大学大学院医学系研究科博士課程 修了,2008年より金沢大学医薬保健研究域医学類 特任助教.
研究テーマ:肝臓に由来する分泌タンパク質“ヘパトカイン”を標的とした新規の2型糖尿病の治療法および診断法の開発.
抱負:ヘパトカインの研究がいつか糖尿病の患者さんを救うことを夢みて日夜邁進しています.
篁 俊成(Toshinari Takamura)
金沢大学医薬保健研究域医学類 准教授.
© 2010 御簾博文・篁 俊成 Licensed under CC 表示 2.1 日本
(金沢大学医薬保健研究域医学類 恒常性制御学)
email:御簾博文,篁 俊成
DOI: 10.7875/first.author.2010.056
A liver-derived secretory protein, selenoprotein P, causes insulin resistance.
Hirofumi Misu, Toshinari Takamura, Hiroaki Takayama, Hiroto Hayashi, Naoto Matsuzawa-Nagata, Seiichiro Kurita, Kazuhide Ishikura, Hitoshi Ando, Yumie Takeshita, Tsuguhito Ota, Masaru Sakurai, Tatsuya Yamashita, Eishiro Mizukoshi, Taro Yamashita, Masao Honda, Ken-ichi Miyamoto, Tetsuya Kubota, Naoto Kubota, Takashi Kadowaki, Han-Jong Kim, In-kyu Lee, Yasuhiko Minokoshi, Yoshiro Saito, Kazuhiko Takahashi, Yoshihiro Yamada, Nobuyuki Takakura, Shuichi Kaneko
Cell Metabolism, 12, 483-495 (2010)
要 約
肝臓はヘパトカインとよばれる分泌タンパク質の産生を介して全身のインスリン感受性を修飾することで糖の恒常性を制御している可能性がある.筆者らは,今回,肝臓に由来する分泌タンパク質のひとつであるselenoprotein Pがインスリン抵抗性の原因となることを明らかにした.まず,ヒト肝臓でのselenoprotein Pの遺伝子発現量がインスリン抵抗性と相関することを見い出した.精製selenoprotein Pによる処理は肝細胞と筋細胞の両方でインスリンシグナルを障害し糖代謝の異常を誘導した.逆に,selenoprotein PのノックアウトならびにRNAiによる抑制ノックダウンはマウスにおいて全身のインスリン感受性と耐糖能を改善した.selenoprotein Pの代謝作用の少なくとも一部はAMPキナーゼの不活性化によるものであった.今回の結果は,selenoprotein Pの糖代謝およびインスリン感受性の制御におけるこれまで知られていなかった役割を明らかにし,selenoprotein Pが2型糖尿病を代表とするインスリン抵抗性関連疾患の治療標的になることを示唆した.
はじめに
インスリン抵抗性は2型糖尿病患者およびメタボリックシンドローム患者の基盤にある病態であり1),心血管系疾患や非アルコール性肝疾患の発症にも関連している.インスリン抵抗性状態ではインスリン作用の障害が肝臓での糖産生の亢進と末梢組織での糖取り込みの低下を誘導し,その結果として高血糖が生じる.インスリン抵抗性の分子機構は完全には理解されていないが,これまで古典的に内分泌臓器と考えられていた臓器とは異なる臓器から分泌される液性因子が,その発症に影響することが知られるようになってきた.たとえば,最近の肥満研究は,脂肪組織はアディポサイトカインとよばれる種々のタンパク質を分泌すること,このアディポサイトカインの産生異常がインスリン抵抗性の発症に寄与することを明らかにしている2).
selenoprotein Pはおもに肝臓で産生される分泌タンパク質である3).10残基のセレノシステイン残基を含んでおり,微量元素であるセレンの輸送タンパク質として機能する4).しかし,その糖代謝ならびにインスリン感受性の制御における役割はこれまで確立されていなかった.さらに,selenoprotein Pノックアウトマウスの研究からその欠損が脳神経障害や不妊と関連することは報告されていたが5,6),ヒト疾患における臨床的な重要性はよくわかっていなかった.
肝臓は糖の恒常性の制御において中心的な役割を担うとともに,多種多様な分泌タンパク質の産生臓器でもある.筆者らは,過去に,2型糖尿病患者の肝臓は分泌タンパク質をコードする膨大な種類の遺伝子を発現していることを報告した7).さらに,2型糖尿病患者の肝臓では血管新生因子,線維化促進因子,酸化ストレス関連因子の発現が健常者とは異なっており8),この変化は2型糖尿病患者の病態形成に貢献していることが推察された.これらの所見を根拠として,脂肪組織と同様に,肝臓が分泌タンパク質の産生を介して2型糖尿病やインスリン抵抗性の発症に寄与しているとの仮説をたてた.このような肝臓に由来する分泌タンパク質をヘパトカインと命名し,インスリン抵抗性関連ヘパトカインの探索を行った(図1).
1.インスリン抵抗性関連ヘパトカインの同定
インスリン抵抗性に関連した肝臓に由来する分泌タンパク質を同定する目的で,ヒト肝生検を行い肝臓に発現する遺伝子を2つの異なった方法を用いて包括的に解析した.まず,2型糖尿病患者5名と健常者5名に対して肝生検を行い,SAGE(serial analysis of gene expression)法を用いて発現遺伝子を包括的に解析した.その結果,分泌タンパク質をコードする遺伝子のうち健常者と比較して2型糖尿病患者で1.5倍以上も発現が変動している117種類の遺伝子を同定した.つぎに,2型糖尿病患者10名と健常者7名に対して超音波ガイドのもとで経皮的な肝生検を行い,DNAチップ解析によって肝臓における遺伝子発現量がインスリン抵抗性と相関する遺伝子を同定した.その結果,SAGE法による解析ではselenoprotein Pの肝臓における遺伝子発現量が2型糖尿病状態では約8倍に上昇していること,DNAチップ解析ではselenoprotein Pの肝臓における遺伝子発現がインスリン感受性と負に相関していることを見い出した.さらに,selenoprotein Pの肝臓における遺伝子発現は2型糖尿病患者の負荷後血糖値あるいは空腹血糖値とも正に相関する傾向を認めた.
2.2型糖尿病状態でのselenoprotein Pの上昇
インスリン抵抗性の発症におけるselenoprotein Pの役割を明らかにするため,ヒト血液中のselenoprotein Pの濃度をELISA法により測定した.肝臓でのselenoprotein Pの遺伝子発現の上昇と一致して,血中selenoprotein P濃度は空腹血糖値とHbA1c値の両方と正に相関していた.HbA1cは過去2~3か月の血糖調節の状態を反映する臨床マーカーである.くわえて,2型糖尿病患者の血中selenoprotein P濃度は健常者と比較して上昇していた.ヒト由来のデータと一致して,2型糖尿病モデル動物であるOLETFラットおよびKKAyマウスにおいても肝臓でのselenoprotein Pの遺伝子発現量と血中濃度は上昇していた.
3.肝細胞でのselenoprotein Pの発現はグルコース,パルミチン酸,インスリンによって制御される
2型糖尿病におけるselenoprotein Pの上昇に寄与する病態を明らかにするため,いくつかの栄養素を肝細胞に処理してselenoprotein Pの遺伝子発現におよぼす影響を検討した.その結果,グルコースおよびパルミチン酸の添加が遺伝子発現を上昇させること,一方,インスリン処理が用量依存的および時間依存的に遺伝子発現を減弱することを見い出した.マウス初代培養肝細胞においてもselenoprotein Pの濃度について同様の結果が認められた.肝細胞におけるインスリンによる負の制御と一致して,空腹状態のマウスの肝臓ではselenoprotein Pの遺伝子発現の上昇が認められた.
4.selenoprotein Pはin vitroにおいてインスリンシグナルを障害し糖代謝の異常を誘導する
selenoprotein Pの遺伝子過剰発現系の作製は技術的に困難をともなうため,クロマトグラフィー法でヒト血液からselenoprotein Pを精製して細胞に対する投与実験を行った.マウス初代肝細胞において精製selenoprotein P処理はインスリン刺激によるインスリン受容体およびその下流のAktのリン酸化を有意に減弱させた.さらに,同様の結果はC2C12筋細胞でも観察された.つぎに,selenoprotein Pが細胞内の糖代謝に異常を起こしうるかどうかを検討した.H4IIEC肝細胞においてselenoprotein Pは糖新生の律速酵素であるPEPCK1およびG6paseの遺伝子発現を上昇させ,その結果,インスリン存在下における肝細胞からの糖放出を30%も増加させた.くわえて,selenoprotein P処理はC2C12筋細胞においてインスリン刺激性の糖取り込みを有意に減弱させた.これらのin vitroでの実験結果は,生理的な濃度でのselenoprotein P処置が細胞のインスリンシグナル伝達を障害し糖代謝の異常を誘導することを明らかにした.
5.selenoprotein Pはin vivoにおいてインスリンシグナルを障害し糖の恒常性の維持をさまたげる
in vivoでのselenoprotein Pの糖代謝におよぼす影響を調べるため,C57BLマウスに精製ヒトselenoprotein Pを実験の12時間前と2時間前の2回,1 mg/kg体重を腹腔内投与した.マウスの血中ヒトselenoprotein P濃度は0.5~1.5μg/mlにいたった.この濃度は健常者と2型糖尿病患者との血中濃度の差分に相当する.糖負荷試験およびインスリン負荷試験は精製selenoprotein Pの投与はマウスに耐糖能の悪化とインスリン抵抗性を誘導することを明らかにした.インスリン誘導性のAktのリン酸化はselenoprotein P投与マウスの肝臓および骨格筋の両方で低下していた.また,精製selenoprotein Pの投与は内因性の糖産生を上昇させ末梢でのグルコース利用率を低下させることが示された.これらの結果は,in vivoにおいてselenoprotein Pが肝臓と骨格筋のインスリンシグナルを障害し耐糖能の異常を誘導することを明らかにした.
6.肝臓でのselenoprotein Pの発現抑制は2型糖尿病マウスの耐糖能の異常とインスリン抵抗性を改善する
内因性のselenoprotein Pのノックダウンがインスリンシグナルを亢進するかどうかを決定するため,H4IIEC肝細胞にselenoprotein Pに特異的なsiRNAを導入した.このselenoprotein Pノックダウン細胞ではインスリン刺激によるAktのリン酸化が有意に増強した.同様に,ハイドロダイナミック法を用いて糖尿病マウスであるKKAyマウスの肝臓にselenoprotein Pに特異的なsiRNAを導入したところ,肝臓および血中のselenoprotein P濃度は約30%も減少した.肝臓でのselenoprotein PのノックダウンはKKAyマウスの耐糖の異常とインスリン抵抗性の両方を改善した.
7.selenoprotein Pノックアウトマウスは良好な耐糖能と肝臓および骨格筋でのインスリンシグナルの亢進を呈する
さらに,selenoprotein P濃度を長期間にわたり低下させることの効果を確認するためselenoprotein Pノックアウトマウスを使用した.selenoprotein Pノックアウトマウスはセレンが十分に含まれた食事で飼育すると生存が可能で正常な体重を呈した.肝臓での脂肪蓄積や脂肪細胞の外観に変化はなかった.しかし,血糖は不変であったが食後インスリン濃度が低値であった.糖負荷試験では耐糖能は良好で,インスリン負荷試験では負荷後血糖は有意に低値であった.インスリンシグナルはselenoprotein Pホモノックアウトマウスの肝臓と骨格筋の両方で増強していた.さらに,selenoprotein Pヘテロノックアウトマウスはインスリン感受性の増強している傾向を認めた.
8.selenoprotein Pの欠損は食事誘導性肥満マウスにおける脂肪細胞の肥大とインスリン抵抗性を軽減する
selenoprotein Pの欠損が食事誘導性肥満によるインスリン抵抗性を減少させるかどうかを明らかにするため,selenoprotein Pノックアウトマウスを高脂肪高ショ糖食で飼育した.体重推移,肝中性脂肪含有量,脂肪組織重量には差を認めなかった.しかし,食事誘導性の脂肪細胞の肥大は軽度であった.空腹時血中インスリン濃度および遊離脂肪酸濃度は有意に低値であった.糖負荷試験およびインスリン負荷試験ではselenoprotein Pホモノックアウトマウスは肥満誘導食での飼育下においても耐糖能の異常とインスリン抵抗性から守られていた.
9.selenoprotein Pは肝細胞でのAMPキナーゼのリン酸化を減少させる
AMPキナーゼはさまざまなエネルギー関連タンパク質をリン酸化するセレンスレオニンキナーゼであり,インスリン感受性を促進するエネルギー制御タンパク質としてはたらく9).selenoprotein P処理はH4IIEC肝細胞とマウス肝臓の両方でAMPキナーゼαとその下流のACCのリン酸化を減弱させることが見い出された.脂肪酸のβ酸化とβ酸化関連遺伝子の発現もselenoprotein P処理で抑制された.selenoprotein Pを処理した肝細胞でAMPおよびATPの濃度は不変であった.機能阻害型AMPキナーゼを肝細胞に導入するとインスリン依存的なAktのリン酸化は減弱し,selenoprotein Pを処理してもこのリン酸化はさらなる低下を認めなかった.逆に,恒常活性型AMPキナーゼを過剰発現させるとselenoprotein Pのインスリン刺激性Aktリン酸化に対する抑制は消失した.これらの結果は,AMPキナーゼのリン酸化の減少がselenoprotein Pによるインスリン抵抗性の誘導の少なくとも一部を仲介することを示唆した.さらに,AMPキナーゼのリン酸化の上流タンパク質の発現を検討した.H4IIEC肝細胞においてAMPキナーゼのリン酸化の負の制御タンパク質であるプロテインホスファターゼ2Cの濃度はselenoprotein P処理によって用量依存的に増加した.
おわりに
今回の研究は,肝臓でのselenoprotein Pの過剰産生が肝臓および骨格筋でのインスリン抵抗性の発症に寄与することを明らかにした(図2).肝臓はおもにグリコーゲンの貯蔵とグルコースの放出を介して糖の恒常性の維持に中心的な役割を担う.くわえて,肝臓は分泌タンパク質の大きな産生源である.ここから,筆者らは,肝臓は“ヘパトカイン”とよぶべき肝臓に由来する分泌タンパク質の産生を介して全身の糖の恒常性を維持するとの仮説をたてた.実際に,過去のいくつかの論文が肝臓に由来する分泌因子とインスリン抵抗性との関連を報告してきた.しかしながら,肝臓に由来しインスリン抵抗性や2型糖尿病の発症に寄与する液性因子の探索はいまだ十分ではないと推測した.なぜならば,筆者らの包括的な遺伝子発現解析では肝臓は分泌タンパク質をコードする膨大な種類の遺伝子を発現しており,その発現は糖尿病状態では変動していたからである7,8).こうして,肝臓における遺伝子発現量とインスリン抵抗性などの臨床パラメーターを比較することでインスリン抵抗性の原因となっている肝臓産生の分泌タンパク質の候補を選択した.この研究は,これまで知られていなかった脂肪組織と同様の肝臓の機能に光をあてた.すなわち,肝臓はホルモンの産生を介してインスリン抵抗性という病態の発症に寄与していたのである.
筆者らの結果は,インスリンが肝細胞におけるselenoprotein Pの発現を負に制御することを示した.この所見と一致して,selenoprotein Pの遺伝子発現はマウスにおいて空腹状態の肝臓で上昇することが示された.空腹状態に代表されるような低インスリン血症の状態では,selenoprotein Pの発現増加は末梢組織での糖取り込みの低下と肝での糖産生を増加させることで低血糖を予防している可能性がある.今回の研究は,肝臓が血液中のインスリン濃度を感知してselenoprotein Pの産生を変動させることで全身のインスリン感受性を制御しているという可能性を提示した.
インスリン標的器官におけるselenoprotein P受容体を同定することがselenoprotein Pによるインスリン抵抗性の誘導機構の解明には必要である.しかしながら,受容体の同定が技術的に困難であったため,今回の研究では糖代謝に作用しうる既知の経路とselenoprotein Pとの関連を探索した.その結果,肝細胞においてselenoprotein PがAMPキナーゼのリン酸化を減弱することを見い出した.さらに,AMPキナーゼリン酸化の負の制御タンパク質であるプロテインホスファターゼ2Cの濃度がselenoprotein Pによって上昇することを実証した.インスリン抵抗性に関連した代表的な炎症性サイトカインであるTNFαもプロテインホスファターゼ2Cの発現増加を介してAMPキナーゼを脱リン酸化することが報告されている10).今後,selenoprotein Pとselenoprotein P受容体との相互作用をさらに検討することで,selenoprotein Pによるプロテインホスファターゼ2Cの上方制御とAMPキナーゼのリン酸化の減少に関するさらなる洞察が得られるものと思われる.
おわりに,今回の研究はインスリン抵抗性および高血糖を誘導する肝臓由来の分泌タンパク質としてselenoprotein Pを同定した.この結果は,ヘパトカインであるselenoprotein Pが2型糖尿病を含むインスリン抵抗性関連疾患の治療標的となりうることを示唆した.
文 献
- Saltiel, A. R. & Kahn, C. R.: Insulin signalling and the regulation of glucose and lipid metabolism. Nature, 414, 799-806 (2001)[PubMed]
- Friedman, J. M. & Halaas, J. L.: Leptin and the regulation of body weight in mammals. Nature, 395, 763-770 (1998)[PubMed]
- Burk, R. F. & Hill, K. E.: Selenoprotein P: an extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu. Rev. Nutr., 25, 215-235 (2005)[PubMed]
- Saito, Y. & Takahashi, K.: Characterization of selenoprotein P as a selenium supply protein. Eur. J. Biochem., 269, 5746-5751 (2002)[PubMed]
- Hill, K. E., Zhou, J., McMahan, W. J. et al.: Deletion of selenoprotein P alters distribution of selenium in the mouse. J. Biol. Chem., 278, 13640-13646 (2003)[PubMed]
- Schomburg, L., Schweizer, U., Holtmann, B. et al.: Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochem. J., 370, 397-402 (2003)[PubMed]
- Misu, H., Takamura, T., Matsuzawa, N. et al.: Genes involved in oxidative phosphorylation are coordinately upregulated with fasting hyperglycaemia in livers of patients with type 2 diabetes. Diabetologia, 50, 268-277 (2007)[PubMed]
- Takamura, T., Sakurai, M., Ota, T. et al.: Genes for systemic vascular complications are differentially expressed in the livers of type 2 diabetic patients. Diabetologia, 47, 638-647 (2004)[PubMed]
- Kahn, B. B., Alquier, T., Carling, D. et al.: AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab., 1, 15-25 (2005)[PubMed]
- Steinberg, G. R., Michell, B. J., van Denderen, B. J. et al.: Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab., 4, 465-474 (2006)[PubMed]
著者プロフィール
略歴:2007年 金沢大学大学院医学系研究科博士課程 修了,2008年より金沢大学医薬保健研究域医学類 特任助教.
研究テーマ:肝臓に由来する分泌タンパク質“ヘパトカイン”を標的とした新規の2型糖尿病の治療法および診断法の開発.
抱負:ヘパトカインの研究がいつか糖尿病の患者さんを救うことを夢みて日夜邁進しています.
篁 俊成(Toshinari Takamura)
金沢大学医薬保健研究域医学類 准教授.
© 2010 御簾博文・篁 俊成 Licensed under CC 表示 2.1 日本
