がん抑制遺伝子Qki遺伝子の欠損によりグリオーマ幹細胞の幹細胞性はニッチの外部においても保持される
新宮多加志・Jian Hu
(米国Texas大学MD Anderson Cancer Center,Department of Cancer Biology)
email:新宮多加志
DOI: 10.7875/first.author.2016.127
Qki deficiency maintains stemness of glioma stem cells in suboptimal environment by downregulating endolysosomal degradation.
Takashi Shingu, Allen L. Ho, Liang Yuan, Xin Zhou, Congxin Dai, Siyuan Zheng, Qianghu Wang, Yi Zhong, Qing Chang, James W. Horner, Brandon D. Liebelt, Yu Yao, Baoli Hu, Yiwen Chen, Gregory N. Fuller, Roeland G. W. Verhaak, Amy B. Heimberger, Jian Hu
Nature Genetics, 49, 75-86 (2017)
ニッチの外部へと遊走し浸潤したがん幹細胞が幹細胞性を保持する機構について十分には明らかにされていない.この研究においては,がん抑制遺伝子のひとつであるQki遺伝子のコンディショナルノックアウトマウスを作製しニッチの外部における神経幹細胞の幹細胞性について検討した.その結果,Qki遺伝子の欠損は神経幹細胞および前がん性の神経幹細胞の自己複製能を亢進し,分化を抑制してニッチの外部において前がん性の神経幹細胞を増加させた.また,マウスにおいてQki遺伝子にくわえPten遺伝子およびTrp53遺伝子を欠損させることによりグリオブラストーマの発生が誘導された.遺伝子発現およびタンパク質量の網羅的な解析などの結果から,Qki遺伝子の欠損によりエンドソーム-リソソーム系が下方制御され,幹細胞性に寄与する細胞膜受容体の取り込みおよび分解が減少し,その結果,前がん性の神経幹細胞の幹細胞性が亢進しニッチの外部においても幹細胞性が保持されると考えられた.この研究は,Qki遺伝子の異常をもつがん幹細胞を標的とするがんの治療法の開発に寄与すると考えられる.
正常な幹細胞はニッチとよばれる至適な環境において自己複製し幹細胞性を維持する1,2).近年,がん組織においてもがん幹細胞に至適な微小環境をもたらすニッチの存在が報告されている3-5).しかし,ニッチからその外部へと遊走,浸潤,転移したがん細胞が幹細胞性を保持する機構について十分には明らかにされていない.
もっとも悪性度の高いグリオーマであるグリオブラストーマは,原発性の悪性の脳腫瘍のうちもっとも頻度が高く,現在の標準的な治療をもってしても転帰および予後は不良である6,7).グリオブラストーマは浸潤性が高く,原発巣から離れた部位へと遊走し浸潤したがん細胞が新たな腫瘍塊を形成する.遊走および浸潤のあいだにさまざまな微小環境を通過するがん細胞が腫瘍の形成能を失わないことから,筆者らは,グリオーマ幹細胞はニッチに依存することなく幹細胞性を保持する能力をもつとの仮説をたてた.
これまで,筆者らは,グリオブラストーマ細胞や前がん性の神経幹細胞において幹細胞性に関与する遺伝子の異常を見い出してきたが,これらの遺伝子のひとつにQki遺伝子がある8,9).その産物であるQkiはSTARファミリーに属するRNA結合タンパク質であり,mRNA前駆体のスプライシング,mRNAの輸送,安定性,翻訳,miRNAのプロセシング,環状RNAの形成などに関与する10).ヒトのグリオブラストーマの検体の25~30%にQki遺伝子の異常が見い出されており,また近年,ほかのがんの研究においてもQki遺伝子はがん抑制遺伝子である可能性が示唆されている7,10).この研究においては,Qki遺伝子のコンディショナルノックアウトマウスおよびそれに由来する神経幹細胞と前がん性の神経幹細胞を用い,Qki遺伝子の欠損がこれらの細胞の幹細胞性,すなわち,自己複製,分化の抑制,腫瘍の形成に及ぼす影響について検討した.
神経幹細胞におけるQki遺伝子の欠損の効果について調べるため,Qki遺伝子のコンディショナルノックアウトマウスを作製し,in vivoにおける効果,とくに,神経幹細胞のニッチである脳室下帯における神経幹細胞の変化について調べた.その結果,Qki遺伝子の欠損により脳室下帯に存在する神経幹細胞の数は対照となるマウスと比べ増加した.Qki遺伝子のコンディショナルノックアウトマウスから分離した神経幹細胞を用いたin vitroにおける解析により,Qki遺伝子の欠損により神経幹細胞の増殖は抑制されるが,自己複製は増強されることが見い出された.また,Qki遺伝子を欠損した神経幹細胞は対照となる神経幹細胞と比較して神経幹細胞のマーカータンパク質の量が多かった.Qki遺伝子の欠損により神経幹細胞の自己複製および神経幹細胞のマーカーの発現が増強されたことから,神経幹細胞の分化が抑制されているかどうかを観察するためQki遺伝子を欠損した神経幹細胞の細胞系譜をin vivoにおいて追跡した.その結果,脳室下帯の外部においてニューロン,アストロサイト,オリゴデンドロサイトへの分化が抑制されていることが確認された.
マウスにおいてQki遺伝子の自然突然変異が報告されているが,このマウスが脳腫瘍を高頻度に発症するという報告はない10).また,予備実験においてQki遺伝子のコンディショナルノックアウトマウスに脳腫瘍は生じなかった.このため,グリオブラストーマにおいて高頻度に異常の認められるがん抑制遺伝子のコンディショナルノックアウトにより,グリオーマの前がん状態を模倣すると考えられるマウスを作製した.その結果,Pten遺伝子およびTrp53遺伝子を欠損したマウスにおいて,脳室下帯の拡大が認められたが脳室下帯の外部において幹細胞性を保持する神経幹細胞はほとんど認められなかった.これに対し,Qki遺伝子,Pten遺伝子,Trp53遺伝子を欠損したマウスにおいては脳室下帯の外部に幹細胞性を保持する神経幹細胞が認められた.また,Pten遺伝子およびTrp53遺伝子を欠損した神経幹細胞とQki遺伝子,Pten遺伝子,Trp53遺伝子を欠損した神経幹細胞とをin vitroにおいて比較したところ,Qki遺伝子のみを欠損した神経幹細胞と同様に,Qki遺伝子の欠損により前がん性の神経幹細胞の増殖は抑制されるが自己複製は増強されることが確認された.さらに,Qki遺伝子,Pten遺伝子,Trp53遺伝子を欠損した神経幹細胞は培養液に含まれるEGFおよびFGFの濃度を標準の1/10としても,なお,神経幹細胞のマーカータンパク質の量がPten遺伝子およびTrp53遺伝子を欠損した神経幹細胞と比較してより多く維持された.これらの所見から,Qki遺伝子の欠損により前がん性の神経幹細胞の幹細胞性は至適の環境ではないニッチの外部においても保持されると考えられた.
Qki遺伝子の欠損によりマウスに脳腫瘍の自然発生がもたらされるかどうかを調べるため,生後7~9日においてPten遺伝子およびTrp53遺伝子の欠損にくわえQki遺伝子を欠損させたところ,92%に脳腫瘍が認められ,これら脳腫瘍においてはアストロサイトのマーカータンパク質の発現およびヒトのグリオブラストーマに特徴的な病理組織学的な所見が認められた.また,遺伝子発現のエンリッチメント解析によりこれらのマウスの脳腫瘍はヒトのグリオブラストーマと同様のサブタイプに分類された.一方,Pten遺伝子およびTrp53遺伝子を欠損したマウスにおいてグリオブラストーマの発生は認められなかった.さらに,Pten遺伝子およびInk4a/Arf遺伝子を欠損したマウスにおいてはグリオブラストーマは認められなかったが,Qki遺伝子,Pten遺伝子,Ink4a/Arf遺伝子を欠損したマウスおいてはグリオブラストーマの発生が認められ,Qki遺伝子の欠損によるグリオブラストーマの発生の促進が確認された.
Qki遺伝子の欠損の効果について神経幹細胞および前がん性の神経幹細胞においてRNAおよびタンパク質につき網羅的に調べたところ,Qkiの直接の標的のうち,mRNAのレベルで104,スプライシングのレベルで73,タンパク質のレベルで148の分子がQkiの欠損により変化していた.これらを用いたパスウェイ解析において,15の経路において集積が認められ,そのうち12の経路は輸送および受容体シグナル伝達系に関するものであった.また,Qki遺伝子の欠損により上方制御されるmRNAの45%,タンパク質の18%は細胞膜受容体あるいは細胞膜への輸送にかかわるものであり,下方制御されるmRNAの39%,タンパク質の40%はエンドサイトーシスあるいはリソソームにかかわるものであった.Qki遺伝子を欠損した神経幹細胞は対照となる神経幹細胞と比較してエンドソームのタンパク質であるRab5およびRab7,リソソームのタンパク質であるLamp1およびLamp2の量が減少していた.さらに,The Cancer Genome Atlasにおけるヒトのグリオブラストーマのデータを解析したところ,Qki遺伝子の発現量とエンドソーム-リソソーム系の遺伝子の発現量とのあいだに正の相関が認められた.これらの結果から,Qkiの減少により細胞膜受容体の細胞膜への輸送は上方制御され,エンドソーム-リソソーム系は下方制御されると考えられた.
Qki遺伝子を欠損した神経幹細胞においては幹細胞性に関連する細胞膜受容体の細胞膜への輸送が亢進し,また,これら細胞膜受容体の取り込みおよび分解が減少して,これらのことが幹細胞性の増強に寄与すると考えた.Pten遺伝子およびTrp53遺伝子を欠損した神経幹細胞においてLamp1をノックダウンしたところ,エンドソーム-リソソーム系の低下による幹細胞性の増強が確認された.幹細胞性に関連する細胞膜受容体について検討したところ,Qki遺伝子のコンディショナルノックアウトマウスの胎仔の脳室下帯においてNotch1,Qki遺伝子のコンディショナルノックアウトマウスの脳室下帯においてFrizzledの発現が亢進しており,また,Qki遺伝子のコンディショナルノックアウトマウスに由来する神経幹細胞においてEGF受容体,Notch1,NICD,TGFβ受容体2,Qki遺伝子,Pten遺伝子,Trp53遺伝子を欠損した神経幹細胞においてWnt5a/bおよびβカテニンのタンパク質量が増加していた.細胞膜受容体の取り込みおよび分解について検討したところ,Qki遺伝子,Pten遺伝子,Trp53遺伝子を欠損した神経幹細胞においては,Pten遺伝子およびTrp53遺伝子を欠損した神経幹細胞と比較してEGF受容体の取り込み,Lamp1とEGF受容体あるいはFrizzled4との共局在が減少していた.また,Qki遺伝子,Pten遺伝子,Trp53遺伝子を欠損した神経幹細胞においてQkiを過剰に発現させるとFGF受容体およびNotch1のタンパク質量が減少した.これらの結果から,Qki遺伝子の欠損により幹細胞性に関連する細胞膜受容体のエンドソーム-リソソーム系による取り込みおよび分解が減少し,このことがQki遺伝子を欠損した神経幹細胞の幹細胞性を増強させ腫瘍原性に寄与することが示唆された.さらに,ヒトのグリオブラストーマの検討においてQkiおよびエンドソーム-リソソーム系のタンパク質量が低い患者では転帰が有意に不良であることが見い出され,ヒトのグリオブラストーマにおけるQkiおよびエンドソーム-リソソーム系の重要性が確認された.
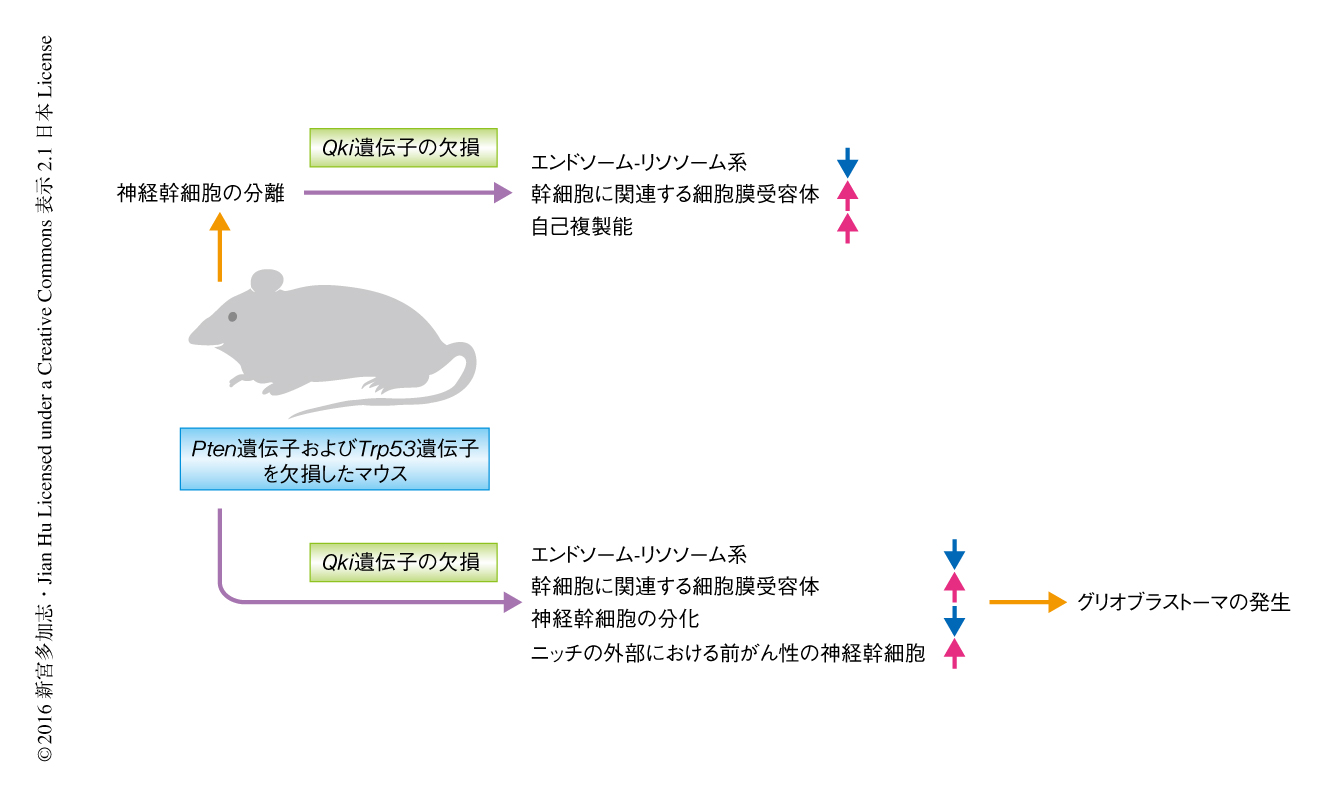
この研究においては,がん細胞が幹細胞性を保持したままニッチから外部へと遊走し浸潤して新たな腫瘍塊を形成する機構を明らかにするため,がん抑制遺伝子のひとつであるQki遺伝子のコンディショナルノックアウトマウスを作製し,これをモデルとして前がん性の神経幹細胞の自己複製,分化,腫瘍原性について検討した.得られた結果から,Qki遺伝子の欠損によりエンドソーム-リソソーム系が下方制御され幹細胞性に寄与する細胞膜受容体の取り込みおよび分解が減少し,その結果,前がん性の神経幹細胞の幹細胞性が亢進しニッチの外部においても幹細胞性が保持され,このことがグリオブラストーマの発生の促進に寄与すると考えられた(図1).この研究は,Qki遺伝子の異常あるいはエンドソーム-リソソーム系の異常をもつがん幹細胞を標的とするがんの治療法の開発に寄与すると考えられる.
略歴:2002年 島根医科大学大学院医学研究科にて博士号取得,2005年 島根大学医学部 講師,2007年 米国Texas大学MD Anderson Cancer Center研究員を経て,2014年より同Instructor.
研究テーマ:Qki遺伝子の欠損によるグリオブラストーマの発生の機序および分子標的治療に対する特異的な脆弱性.
関心事:グリオーマの発生の機序の解明および治療法の開発.
Jian Hu
米国Texas大学MD Anderson Cancer CenterにてAssistant Professor.
© 2016 新宮多加志・Jian Hu Licensed under CC 表示 2.1 日本
(米国Texas大学MD Anderson Cancer Center,Department of Cancer Biology)
email:新宮多加志
DOI: 10.7875/first.author.2016.127
Qki deficiency maintains stemness of glioma stem cells in suboptimal environment by downregulating endolysosomal degradation.
Takashi Shingu, Allen L. Ho, Liang Yuan, Xin Zhou, Congxin Dai, Siyuan Zheng, Qianghu Wang, Yi Zhong, Qing Chang, James W. Horner, Brandon D. Liebelt, Yu Yao, Baoli Hu, Yiwen Chen, Gregory N. Fuller, Roeland G. W. Verhaak, Amy B. Heimberger, Jian Hu
Nature Genetics, 49, 75-86 (2017)
要 約
ニッチの外部へと遊走し浸潤したがん幹細胞が幹細胞性を保持する機構について十分には明らかにされていない.この研究においては,がん抑制遺伝子のひとつであるQki遺伝子のコンディショナルノックアウトマウスを作製しニッチの外部における神経幹細胞の幹細胞性について検討した.その結果,Qki遺伝子の欠損は神経幹細胞および前がん性の神経幹細胞の自己複製能を亢進し,分化を抑制してニッチの外部において前がん性の神経幹細胞を増加させた.また,マウスにおいてQki遺伝子にくわえPten遺伝子およびTrp53遺伝子を欠損させることによりグリオブラストーマの発生が誘導された.遺伝子発現およびタンパク質量の網羅的な解析などの結果から,Qki遺伝子の欠損によりエンドソーム-リソソーム系が下方制御され,幹細胞性に寄与する細胞膜受容体の取り込みおよび分解が減少し,その結果,前がん性の神経幹細胞の幹細胞性が亢進しニッチの外部においても幹細胞性が保持されると考えられた.この研究は,Qki遺伝子の異常をもつがん幹細胞を標的とするがんの治療法の開発に寄与すると考えられる.
はじめに
正常な幹細胞はニッチとよばれる至適な環境において自己複製し幹細胞性を維持する1,2).近年,がん組織においてもがん幹細胞に至適な微小環境をもたらすニッチの存在が報告されている3-5).しかし,ニッチからその外部へと遊走,浸潤,転移したがん細胞が幹細胞性を保持する機構について十分には明らかにされていない.
もっとも悪性度の高いグリオーマであるグリオブラストーマは,原発性の悪性の脳腫瘍のうちもっとも頻度が高く,現在の標準的な治療をもってしても転帰および予後は不良である6,7).グリオブラストーマは浸潤性が高く,原発巣から離れた部位へと遊走し浸潤したがん細胞が新たな腫瘍塊を形成する.遊走および浸潤のあいだにさまざまな微小環境を通過するがん細胞が腫瘍の形成能を失わないことから,筆者らは,グリオーマ幹細胞はニッチに依存することなく幹細胞性を保持する能力をもつとの仮説をたてた.
これまで,筆者らは,グリオブラストーマ細胞や前がん性の神経幹細胞において幹細胞性に関与する遺伝子の異常を見い出してきたが,これらの遺伝子のひとつにQki遺伝子がある8,9).その産物であるQkiはSTARファミリーに属するRNA結合タンパク質であり,mRNA前駆体のスプライシング,mRNAの輸送,安定性,翻訳,miRNAのプロセシング,環状RNAの形成などに関与する10).ヒトのグリオブラストーマの検体の25~30%にQki遺伝子の異常が見い出されており,また近年,ほかのがんの研究においてもQki遺伝子はがん抑制遺伝子である可能性が示唆されている7,10).この研究においては,Qki遺伝子のコンディショナルノックアウトマウスおよびそれに由来する神経幹細胞と前がん性の神経幹細胞を用い,Qki遺伝子の欠損がこれらの細胞の幹細胞性,すなわち,自己複製,分化の抑制,腫瘍の形成に及ぼす影響について検討した.
1.Qki遺伝子の欠損は神経幹細胞の自己複製を増強し分化を抑制する
神経幹細胞におけるQki遺伝子の欠損の効果について調べるため,Qki遺伝子のコンディショナルノックアウトマウスを作製し,in vivoにおける効果,とくに,神経幹細胞のニッチである脳室下帯における神経幹細胞の変化について調べた.その結果,Qki遺伝子の欠損により脳室下帯に存在する神経幹細胞の数は対照となるマウスと比べ増加した.Qki遺伝子のコンディショナルノックアウトマウスから分離した神経幹細胞を用いたin vitroにおける解析により,Qki遺伝子の欠損により神経幹細胞の増殖は抑制されるが,自己複製は増強されることが見い出された.また,Qki遺伝子を欠損した神経幹細胞は対照となる神経幹細胞と比較して神経幹細胞のマーカータンパク質の量が多かった.Qki遺伝子の欠損により神経幹細胞の自己複製および神経幹細胞のマーカーの発現が増強されたことから,神経幹細胞の分化が抑制されているかどうかを観察するためQki遺伝子を欠損した神経幹細胞の細胞系譜をin vivoにおいて追跡した.その結果,脳室下帯の外部においてニューロン,アストロサイト,オリゴデンドロサイトへの分化が抑制されていることが確認された.
2.Qki遺伝子の欠損により前がん性の神経幹細胞の幹細胞性はニッチの外部においても保持される
マウスにおいてQki遺伝子の自然突然変異が報告されているが,このマウスが脳腫瘍を高頻度に発症するという報告はない10).また,予備実験においてQki遺伝子のコンディショナルノックアウトマウスに脳腫瘍は生じなかった.このため,グリオブラストーマにおいて高頻度に異常の認められるがん抑制遺伝子のコンディショナルノックアウトにより,グリオーマの前がん状態を模倣すると考えられるマウスを作製した.その結果,Pten遺伝子およびTrp53遺伝子を欠損したマウスにおいて,脳室下帯の拡大が認められたが脳室下帯の外部において幹細胞性を保持する神経幹細胞はほとんど認められなかった.これに対し,Qki遺伝子,Pten遺伝子,Trp53遺伝子を欠損したマウスにおいては脳室下帯の外部に幹細胞性を保持する神経幹細胞が認められた.また,Pten遺伝子およびTrp53遺伝子を欠損した神経幹細胞とQki遺伝子,Pten遺伝子,Trp53遺伝子を欠損した神経幹細胞とをin vitroにおいて比較したところ,Qki遺伝子のみを欠損した神経幹細胞と同様に,Qki遺伝子の欠損により前がん性の神経幹細胞の増殖は抑制されるが自己複製は増強されることが確認された.さらに,Qki遺伝子,Pten遺伝子,Trp53遺伝子を欠損した神経幹細胞は培養液に含まれるEGFおよびFGFの濃度を標準の1/10としても,なお,神経幹細胞のマーカータンパク質の量がPten遺伝子およびTrp53遺伝子を欠損した神経幹細胞と比較してより多く維持された.これらの所見から,Qki遺伝子の欠損により前がん性の神経幹細胞の幹細胞性は至適の環境ではないニッチの外部においても保持されると考えられた.
3.Pten遺伝子およびTrp53遺伝子の欠損にともなうQki遺伝子の欠損はマウスにおいてグリオブラストーマの発生を促進する
Qki遺伝子の欠損によりマウスに脳腫瘍の自然発生がもたらされるかどうかを調べるため,生後7~9日においてPten遺伝子およびTrp53遺伝子の欠損にくわえQki遺伝子を欠損させたところ,92%に脳腫瘍が認められ,これら脳腫瘍においてはアストロサイトのマーカータンパク質の発現およびヒトのグリオブラストーマに特徴的な病理組織学的な所見が認められた.また,遺伝子発現のエンリッチメント解析によりこれらのマウスの脳腫瘍はヒトのグリオブラストーマと同様のサブタイプに分類された.一方,Pten遺伝子およびTrp53遺伝子を欠損したマウスにおいてグリオブラストーマの発生は認められなかった.さらに,Pten遺伝子およびInk4a/Arf遺伝子を欠損したマウスにおいてはグリオブラストーマは認められなかったが,Qki遺伝子,Pten遺伝子,Ink4a/Arf遺伝子を欠損したマウスおいてはグリオブラストーマの発生が認められ,Qki遺伝子の欠損によるグリオブラストーマの発生の促進が確認された.
4.Qkiはエンドソーム-リソソーム系を制御する
Qki遺伝子の欠損の効果について神経幹細胞および前がん性の神経幹細胞においてRNAおよびタンパク質につき網羅的に調べたところ,Qkiの直接の標的のうち,mRNAのレベルで104,スプライシングのレベルで73,タンパク質のレベルで148の分子がQkiの欠損により変化していた.これらを用いたパスウェイ解析において,15の経路において集積が認められ,そのうち12の経路は輸送および受容体シグナル伝達系に関するものであった.また,Qki遺伝子の欠損により上方制御されるmRNAの45%,タンパク質の18%は細胞膜受容体あるいは細胞膜への輸送にかかわるものであり,下方制御されるmRNAの39%,タンパク質の40%はエンドサイトーシスあるいはリソソームにかかわるものであった.Qki遺伝子を欠損した神経幹細胞は対照となる神経幹細胞と比較してエンドソームのタンパク質であるRab5およびRab7,リソソームのタンパク質であるLamp1およびLamp2の量が減少していた.さらに,The Cancer Genome Atlasにおけるヒトのグリオブラストーマのデータを解析したところ,Qki遺伝子の発現量とエンドソーム-リソソーム系の遺伝子の発現量とのあいだに正の相関が認められた.これらの結果から,Qkiの減少により細胞膜受容体の細胞膜への輸送は上方制御され,エンドソーム-リソソーム系は下方制御されると考えられた.
5.Qki遺伝子の欠損により細胞膜受容体の分解が減少し前がん性の神経幹細胞の幹細胞性が増強する
Qki遺伝子を欠損した神経幹細胞においては幹細胞性に関連する細胞膜受容体の細胞膜への輸送が亢進し,また,これら細胞膜受容体の取り込みおよび分解が減少して,これらのことが幹細胞性の増強に寄与すると考えた.Pten遺伝子およびTrp53遺伝子を欠損した神経幹細胞においてLamp1をノックダウンしたところ,エンドソーム-リソソーム系の低下による幹細胞性の増強が確認された.幹細胞性に関連する細胞膜受容体について検討したところ,Qki遺伝子のコンディショナルノックアウトマウスの胎仔の脳室下帯においてNotch1,Qki遺伝子のコンディショナルノックアウトマウスの脳室下帯においてFrizzledの発現が亢進しており,また,Qki遺伝子のコンディショナルノックアウトマウスに由来する神経幹細胞においてEGF受容体,Notch1,NICD,TGFβ受容体2,Qki遺伝子,Pten遺伝子,Trp53遺伝子を欠損した神経幹細胞においてWnt5a/bおよびβカテニンのタンパク質量が増加していた.細胞膜受容体の取り込みおよび分解について検討したところ,Qki遺伝子,Pten遺伝子,Trp53遺伝子を欠損した神経幹細胞においては,Pten遺伝子およびTrp53遺伝子を欠損した神経幹細胞と比較してEGF受容体の取り込み,Lamp1とEGF受容体あるいはFrizzled4との共局在が減少していた.また,Qki遺伝子,Pten遺伝子,Trp53遺伝子を欠損した神経幹細胞においてQkiを過剰に発現させるとFGF受容体およびNotch1のタンパク質量が減少した.これらの結果から,Qki遺伝子の欠損により幹細胞性に関連する細胞膜受容体のエンドソーム-リソソーム系による取り込みおよび分解が減少し,このことがQki遺伝子を欠損した神経幹細胞の幹細胞性を増強させ腫瘍原性に寄与することが示唆された.さらに,ヒトのグリオブラストーマの検討においてQkiおよびエンドソーム-リソソーム系のタンパク質量が低い患者では転帰が有意に不良であることが見い出され,ヒトのグリオブラストーマにおけるQkiおよびエンドソーム-リソソーム系の重要性が確認された.
おわりに
この研究においては,がん細胞が幹細胞性を保持したままニッチから外部へと遊走し浸潤して新たな腫瘍塊を形成する機構を明らかにするため,がん抑制遺伝子のひとつであるQki遺伝子のコンディショナルノックアウトマウスを作製し,これをモデルとして前がん性の神経幹細胞の自己複製,分化,腫瘍原性について検討した.得られた結果から,Qki遺伝子の欠損によりエンドソーム-リソソーム系が下方制御され幹細胞性に寄与する細胞膜受容体の取り込みおよび分解が減少し,その結果,前がん性の神経幹細胞の幹細胞性が亢進しニッチの外部においても幹細胞性が保持され,このことがグリオブラストーマの発生の促進に寄与すると考えられた(図1).この研究は,Qki遺伝子の異常あるいはエンドソーム-リソソーム系の異常をもつがん幹細胞を標的とするがんの治療法の開発に寄与すると考えられる.
文 献
- He, S., Nakada, D. & Morrison, S. J.: Mechanisms of stem cell self-renewal. Annu. Rev. Cell Dev. Biol., 25, 377-406 (2009)[PubMed]
- Ming, G. L. & Song, H.: Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron, 70, 687-702 (2011)[PubMed]
- Calabrese, C., Poppleton, H., Kocak. M. et al.: A perivascular niche for brain tumor stem cells. Cancer Cell, 11, 69-82 (2007)[PubMed]
- Lane, S. W., Scadden, D. T. & Gilliland, D. G.: The leukemic stem cell niche: current concepts and therapeutic opportunities. Blood, 114, 1150-1157 (2009)[PubMed]
- Li, L. & Neaves, W. B.: Normal stem cells and cancer stem cells: the niche matters. Cancer Res., 66, 4553-4557 (2006)[PubMed]
- Stupp, R., Mason. W. P., van den Bent., M. J. et al.: Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med., 352, 987-996 (2005)[PubMed]
- Brennan, C. W., Verhaak., R. G. W., McKenna. A. et al.: The somatic genomic landscape of glioblastoma. Cell, 155, 462-477 (2013)[PubMed]
- Hu, J., Ho, A. L., Hu, B. et al.: Neutralization of terminal differentiation in gliomagenesis. Proc. Natl. Acad. Sci. USA, 110, 14520-14527 (2013)[PubMed]
- Chen, A. J., Paik, J. H., Zhang, H. et al.: STAR RNA-binding protein Quaking suppresses cancer via stabilization of specific miRNA. Genes Dev., 26, 1459-1472 (2012)[PubMed]
- Darbelli, L. & Richard, S.: Emerging functions of the Quaking RNA-binding proteins and link to human diseases. Wiley Interdiscip. Rev. RNA, 7, 399-412 (2016)[PubMed]
活用したデータベースにかかわるキーワードと統合TVへのリンク
著者プロフィール
略歴:2002年 島根医科大学大学院医学研究科にて博士号取得,2005年 島根大学医学部 講師,2007年 米国Texas大学MD Anderson Cancer Center研究員を経て,2014年より同Instructor.
研究テーマ:Qki遺伝子の欠損によるグリオブラストーマの発生の機序および分子標的治療に対する特異的な脆弱性.
関心事:グリオーマの発生の機序の解明および治療法の開発.
Jian Hu
米国Texas大学MD Anderson Cancer CenterにてAssistant Professor.
© 2016 新宮多加志・Jian Hu Licensed under CC 表示 2.1 日本
