次世代シークエンサーによる日本人全ゲノム配列のはじめての包括的な解析
角田 達彦
(理化学研究所ゲノム医科学研究センター 情報解析研究チーム)
email:角田達彦
DOI: 10.7875/first.author.2010.049
Whole-genome sequencing and comprehensive variant analysis of a Japanese individual using massively parallel sequencing.
Akihiro Fujimoto, Hidewaki Nakagawa, Naoya Hosono, Kaoru Nakano, Tetsuo Abe, Keith A Boroevich, Masao Nagasaki, Rui Yamaguchi, Tetsuo Shibuya, Michiaki Kubo, Satoru Miyano, Yusuke Nakamura, Tatsuhiko Tsunoda
Nature Genetics, 42, 931-936 (2010)
今回,筆者らは,次世代シークエンサーを使い日本人1人の全ゲノム配列の決定と高精度な解析を行った.日本人の全ゲノム配列が報告されたのははじめてである.取得したデータの99%以上は既知のヒトゲノム参照配列にマップすることができた.このデータにベイズ決定法を適用することで約313万個の一塩基多様性を約99.9%の高精度で検出した.そして,海外の研究グループからの6人の全ゲノム配列と日本人の全ゲノム配列とを比較し,集団では見失われていた,遺伝子の機能に影響をあたえるであろう一塩基多様性が個人個人には多いことを発見した.また,高精度な方法により10 kbpより小さい欠失を約5300個検出し,コピー数の多様性や構造の多様性も網羅的に見い出し,さらに,ヒトゲノム参照配列にない約3 Mbの新規配列を発見した.ヒトゲノムには多様性に富むDNA塩基配列がまだまだ数多く存在し,全ゲノム配列の解析はそれらを完全に理解するのにきわめて重要なアプローチである.今後,同じ方法により日本人全ゲノム配列の多様性が解析され,日本人のための疾患研究の展開することが期待される.
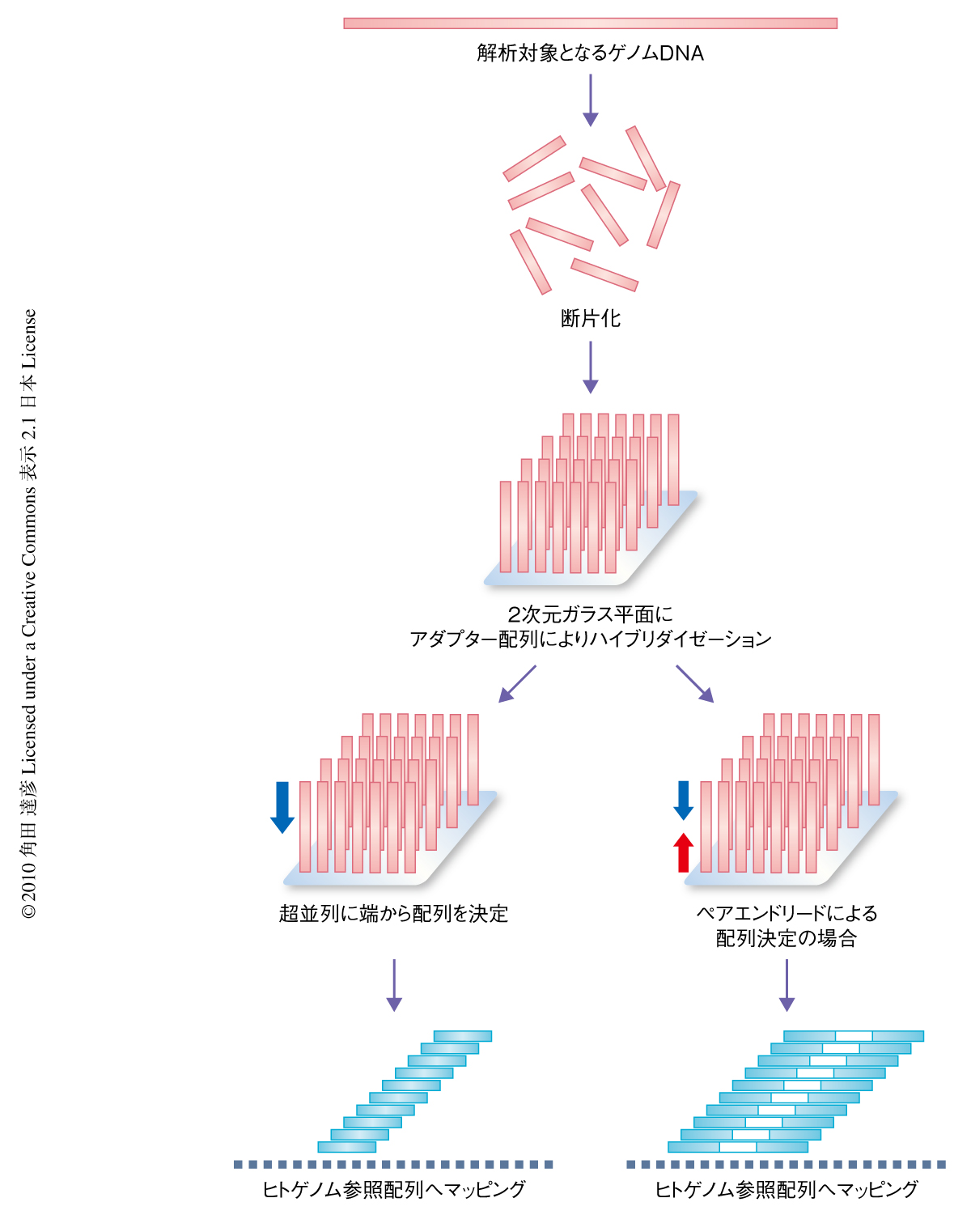
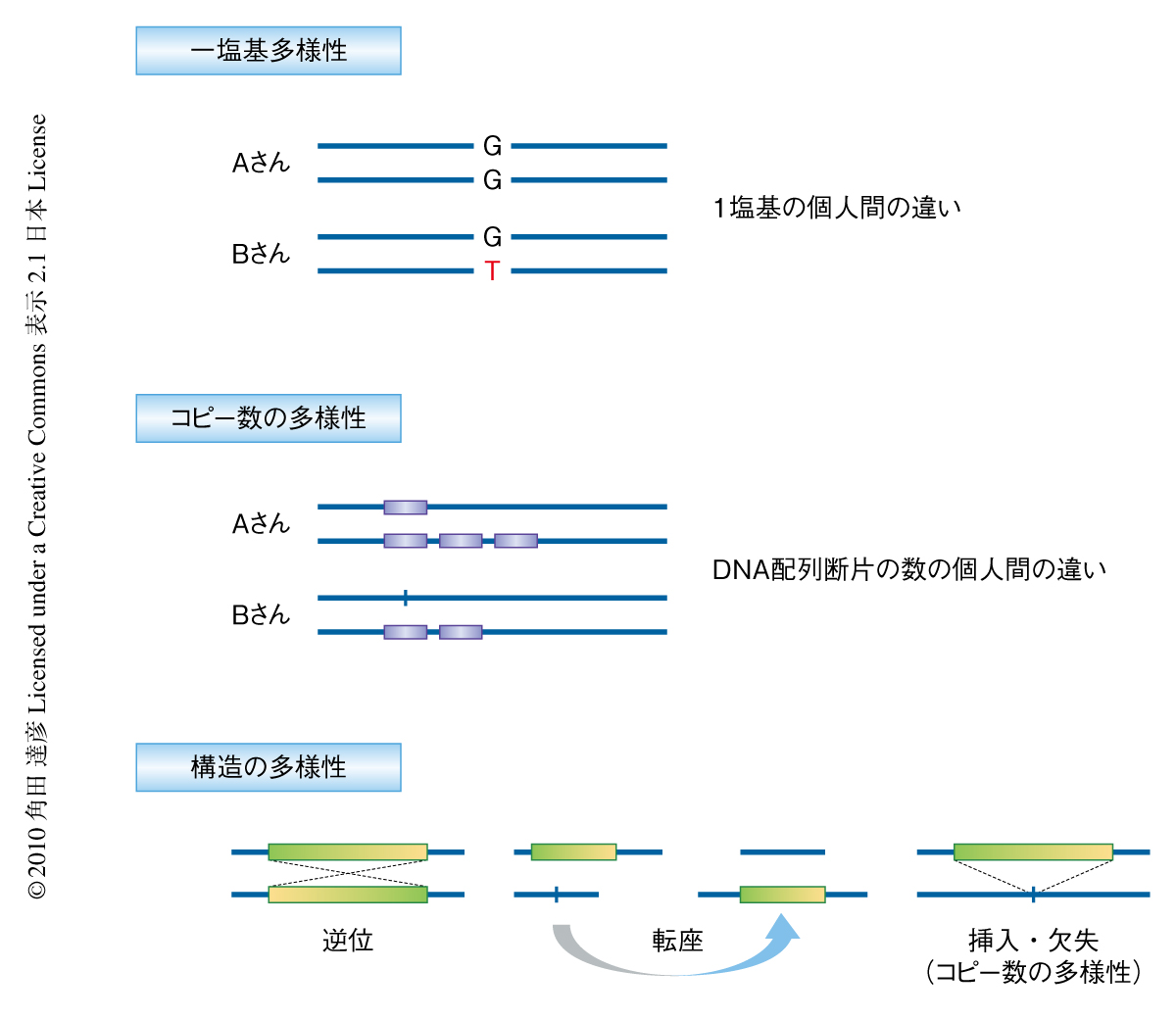
現在,筆者らのグループをはじめ全世界で,タイピングチップを用いて患者グループと対照グループとで一塩基多型(single nucleotide polymorphism:SNP)の対立遺伝子を比較して疾患に関連する遺伝子を探索するゲノムワイド関連解析という方法が爆発的に行われるようになり,医学がたいへんな勢いで進展している1,2).しかし,このゲノムワイド関連解析は基本的には集団内で比較的多くの人が共通してもつような(対立遺伝子の頻度が5%以上)SNPのセットを用いており,より低い頻度をもつ多様性を解明するにはさらなるくふうが必要である3-8).そこで,個人の全ゲノム配列の解析が行われるようになってきた9).これには,次世代シークエンサーとよばれる超並列で塩基配列の決定を行う機器・技術(図1)が急速に発展しつつあり低コストかつ短時間で配列決定のできるようになってきたという背景も後押ししている.そして,その技術を使うことにより,一塩基多様性,コピー数の多様性,挿入・欠失や転座など構造の多様性(図2)を検出するための情報がいちどに同じプラットフォームから得られることもわかってきた.しかし,その技術は,まだ読み取り断片の長さが短い(50 bpから150 bp程度まで),1回あたりの読み取りエラーが比較的高い,といった問題があり,ほかにも,マッピングのエラー,ヒトゲノム参照ゲノム配列が日本人にどれくらい使えるのか,検出アルゴリズムの違いによる影響が大きい,などといった問題もある.これらの問題を解決するためには,塩基配列の決定技術の改良とともに,より洗練された情報科学的なアプローチが必要である.また,これまでは日本人の全ゲノム配列の包括的な解析もなされておらず,固有の配列や多様性の有無,描像なども不明であった.
筆者らは,米国Illumina社のGenome Analyzer IIという次世代シークエンサーを用いて,国際HapMapプロジェクト10) で解析された日本人男性1人のDNAから全部で約120 Gbpのデータを得た.精度をよくするためすべての配列を約40回ずつ解読している.その99%以上は米国NCBI(National Center for Biotechnology Information)に登録されているヒトゲノム参照配列にマップすることができた.一塩基多様性の検出のため,カウント法,頻度法,ベイズ決定法による結果を比較し,もっとも高精度だったベイズ決定法を用いることにした.その結果,3,132,608個の一塩基多様性が得られた.従来のSNP解析用のDNAチップにも搭載されていた一塩基多様性についてDNAチップによる結果と比較すると約99.9%は一致し,塩基配列の解析結果が高精度であることを確認できた.また,今回,検出した一塩基多様性のうち12.6%にあたる395,940個は既知のデータベースにはなく新規のものだった.さらに,タンパク質コード領域内で,9783個のミスセンス一塩基多様性と,96個のナンセンス一塩基多様性を見い出した.そのうえ,217,176個の短い配列の挿入や228,063個の短い配列の欠失を検出したが,そのうちの487個はタンパク質コード領域内に存在していた.タンパク質コード領域内には351個の3文字単位でない塩基の挿入や欠失も見い出された.これらは遺伝子の機能に大きな影響をあたえている可能性がある.
今回の日本人の全ゲノム配列にくわえて,海外の研究から得られていた欧米人,アフリカ人,中国人,韓国人の6人の全ゲノム配列の一塩基多様性のデータをあわせて解析した.すると,個人個人には集団では見失われていたミスセンス一塩基多様性やナンセンス一塩基多様性の多いことがわかった.すなわち,遺伝子機能によくない影響をあたえるであろう一塩基多様性のほとんどは自然選択によりまれなものとなるため,これまでの集団内でのSNPの探索では大多数が見失われてきたものと推測できた.また,遺伝子の機能別に分類して解析するとナンセンス一塩基多様性は嗅覚や化学的な刺激の認識に関係するものの多いことがわかった.
配列の欠失の検出についてはデータリードの深さとリード対のあいだの距離との両方を使う高精度な方法を実現した.その結果,5319個の欠失が候補として検出された.それらの一部をPCR法で検証するとすべて正解であった.この方法を使うと従来のDNAチップの技術では検出のむずかしいかった数百bpの小さな欠失を検出することが可能となる.また,検出した欠失のうち,74個が70個の遺伝子領域(126個のエキソン)と重なることがわかった.これらは遺伝子の機能に影響をあたえている可能性がある.
10 kbp以上という長い配列のコピー数の多様性の検出には5 kbpの範囲内の配列の解読された回数を情報として使用した.その結果,コピー数の多い領域113個と,コピー数の少ない領域109個を検出した.それらを別の実験により検証したところ結果のよく一致することがわかった.この技術の大きな特徴はほかのサンプルと比較することなく1サンプルだけで検出の可能なことである.また,57個の逆位,112個の染色体内転座の候補も見い出された.
塩基配列のアセンブリーを行うABySS,SOAPdenovo,Velvetという3種類のソフトウェアを使ってヒトゲノム参照配列にマップできなかったデータをアセンブリーした結果,それぞれ6535個,4826個,6617個のコンティグが得られた.3つのソフトウェアが出す結果はたがいによく似ており,コンティグのうち185個をPCR法で検証すると181個が実際に存在することがわかった.さらに,新規配列の90%以上は通常の塩基配列の解析法でも確認できた.全部で3~3.4 Mbpがヒトゲノム参照配列にない新規の配列だった.
一般に,有害な遺伝的多様性は自然選択のため集団内では広がらない方向に抑えられているものと考えられるが,個人のDNAのレベルでは疾患関連の多様性がまだまだ多くみつかる可能性がある.全ゲノム配列の解析は,そのような集団内の頻度の低い多様性を網羅的に検出する本質的な技術となりうる.全ゲノム配列の解析のもうひとつの特徴は新規の配列を発見できることである.2003年に配列決定が完了したとされ,現在,多くの研究者が用いているヒトゲノム参照配列のみを用いることでは不十分であろう.今後,より多くの日本人の全ゲノム配列を決定することにより,日本人のための研究の基盤づくりが期待されるとともに,全ゲノム配列の解析によっていろいろな疾患に関連する未知の多様性が発見されるものと考えられる.また,今回の解析技術をすでに開始しているがんゲノム解析を行う国際がんゲノムコンソーシアム(International Cancer Genome Consortium:ICGC)で活用することにより,がんゲノムの包括的な情報を解明しがんの原因にせまることができるであろう.これらの結果,オーダーメイド医療がますます進展し,医学および医療に大きな展開をもたらすことになろう.
(理化学研究所ゲノム医科学研究センター 情報解析研究チーム)
email:角田達彦
DOI: 10.7875/first.author.2010.049
Whole-genome sequencing and comprehensive variant analysis of a Japanese individual using massively parallel sequencing.
Akihiro Fujimoto, Hidewaki Nakagawa, Naoya Hosono, Kaoru Nakano, Tetsuo Abe, Keith A Boroevich, Masao Nagasaki, Rui Yamaguchi, Tetsuo Shibuya, Michiaki Kubo, Satoru Miyano, Yusuke Nakamura, Tatsuhiko Tsunoda
Nature Genetics, 42, 931-936 (2010)
要 約
今回,筆者らは,次世代シークエンサーを使い日本人1人の全ゲノム配列の決定と高精度な解析を行った.日本人の全ゲノム配列が報告されたのははじめてである.取得したデータの99%以上は既知のヒトゲノム参照配列にマップすることができた.このデータにベイズ決定法を適用することで約313万個の一塩基多様性を約99.9%の高精度で検出した.そして,海外の研究グループからの6人の全ゲノム配列と日本人の全ゲノム配列とを比較し,集団では見失われていた,遺伝子の機能に影響をあたえるであろう一塩基多様性が個人個人には多いことを発見した.また,高精度な方法により10 kbpより小さい欠失を約5300個検出し,コピー数の多様性や構造の多様性も網羅的に見い出し,さらに,ヒトゲノム参照配列にない約3 Mbの新規配列を発見した.ヒトゲノムには多様性に富むDNA塩基配列がまだまだ数多く存在し,全ゲノム配列の解析はそれらを完全に理解するのにきわめて重要なアプローチである.今後,同じ方法により日本人全ゲノム配列の多様性が解析され,日本人のための疾患研究の展開することが期待される.
はじめに
現在,筆者らのグループをはじめ全世界で,タイピングチップを用いて患者グループと対照グループとで一塩基多型(single nucleotide polymorphism:SNP)の対立遺伝子を比較して疾患に関連する遺伝子を探索するゲノムワイド関連解析という方法が爆発的に行われるようになり,医学がたいへんな勢いで進展している1,2).しかし,このゲノムワイド関連解析は基本的には集団内で比較的多くの人が共通してもつような(対立遺伝子の頻度が5%以上)SNPのセットを用いており,より低い頻度をもつ多様性を解明するにはさらなるくふうが必要である3-8).そこで,個人の全ゲノム配列の解析が行われるようになってきた9).これには,次世代シークエンサーとよばれる超並列で塩基配列の決定を行う機器・技術(図1)が急速に発展しつつあり低コストかつ短時間で配列決定のできるようになってきたという背景も後押ししている.そして,その技術を使うことにより,一塩基多様性,コピー数の多様性,挿入・欠失や転座など構造の多様性(図2)を検出するための情報がいちどに同じプラットフォームから得られることもわかってきた.しかし,その技術は,まだ読み取り断片の長さが短い(50 bpから150 bp程度まで),1回あたりの読み取りエラーが比較的高い,といった問題があり,ほかにも,マッピングのエラー,ヒトゲノム参照ゲノム配列が日本人にどれくらい使えるのか,検出アルゴリズムの違いによる影響が大きい,などといった問題もある.これらの問題を解決するためには,塩基配列の決定技術の改良とともに,より洗練された情報科学的なアプローチが必要である.また,これまでは日本人の全ゲノム配列の包括的な解析もなされておらず,固有の配列や多様性の有無,描像なども不明であった.
1.一塩基多様性の検出
筆者らは,米国Illumina社のGenome Analyzer IIという次世代シークエンサーを用いて,国際HapMapプロジェクト10) で解析された日本人男性1人のDNAから全部で約120 Gbpのデータを得た.精度をよくするためすべての配列を約40回ずつ解読している.その99%以上は米国NCBI(National Center for Biotechnology Information)に登録されているヒトゲノム参照配列にマップすることができた.一塩基多様性の検出のため,カウント法,頻度法,ベイズ決定法による結果を比較し,もっとも高精度だったベイズ決定法を用いることにした.その結果,3,132,608個の一塩基多様性が得られた.従来のSNP解析用のDNAチップにも搭載されていた一塩基多様性についてDNAチップによる結果と比較すると約99.9%は一致し,塩基配列の解析結果が高精度であることを確認できた.また,今回,検出した一塩基多様性のうち12.6%にあたる395,940個は既知のデータベースにはなく新規のものだった.さらに,タンパク質コード領域内で,9783個のミスセンス一塩基多様性と,96個のナンセンス一塩基多様性を見い出した.そのうえ,217,176個の短い配列の挿入や228,063個の短い配列の欠失を検出したが,そのうちの487個はタンパク質コード領域内に存在していた.タンパク質コード領域内には351個の3文字単位でない塩基の挿入や欠失も見い出された.これらは遺伝子の機能に大きな影響をあたえている可能性がある.
2.7人のゲノムの統合比較解析
今回の日本人の全ゲノム配列にくわえて,海外の研究から得られていた欧米人,アフリカ人,中国人,韓国人の6人の全ゲノム配列の一塩基多様性のデータをあわせて解析した.すると,個人個人には集団では見失われていたミスセンス一塩基多様性やナンセンス一塩基多様性の多いことがわかった.すなわち,遺伝子機能によくない影響をあたえるであろう一塩基多様性のほとんどは自然選択によりまれなものとなるため,これまでの集団内でのSNPの探索では大多数が見失われてきたものと推測できた.また,遺伝子の機能別に分類して解析するとナンセンス一塩基多様性は嗅覚や化学的な刺激の認識に関係するものの多いことがわかった.
3.配列の欠失の検出
配列の欠失の検出についてはデータリードの深さとリード対のあいだの距離との両方を使う高精度な方法を実現した.その結果,5319個の欠失が候補として検出された.それらの一部をPCR法で検証するとすべて正解であった.この方法を使うと従来のDNAチップの技術では検出のむずかしいかった数百bpの小さな欠失を検出することが可能となる.また,検出した欠失のうち,74個が70個の遺伝子領域(126個のエキソン)と重なることがわかった.これらは遺伝子の機能に影響をあたえている可能性がある.
4.長い配列のコピー数の多様性の検出
10 kbp以上という長い配列のコピー数の多様性の検出には5 kbpの範囲内の配列の解読された回数を情報として使用した.その結果,コピー数の多い領域113個と,コピー数の少ない領域109個を検出した.それらを別の実験により検証したところ結果のよく一致することがわかった.この技術の大きな特徴はほかのサンプルと比較することなく1サンプルだけで検出の可能なことである.また,57個の逆位,112個の染色体内転座の候補も見い出された.
5.新規配列の検出
塩基配列のアセンブリーを行うABySS,SOAPdenovo,Velvetという3種類のソフトウェアを使ってヒトゲノム参照配列にマップできなかったデータをアセンブリーした結果,それぞれ6535個,4826個,6617個のコンティグが得られた.3つのソフトウェアが出す結果はたがいによく似ており,コンティグのうち185個をPCR法で検証すると181個が実際に存在することがわかった.さらに,新規配列の90%以上は通常の塩基配列の解析法でも確認できた.全部で3~3.4 Mbpがヒトゲノム参照配列にない新規の配列だった.
おわりに
一般に,有害な遺伝的多様性は自然選択のため集団内では広がらない方向に抑えられているものと考えられるが,個人のDNAのレベルでは疾患関連の多様性がまだまだ多くみつかる可能性がある.全ゲノム配列の解析は,そのような集団内の頻度の低い多様性を網羅的に検出する本質的な技術となりうる.全ゲノム配列の解析のもうひとつの特徴は新規の配列を発見できることである.2003年に配列決定が完了したとされ,現在,多くの研究者が用いているヒトゲノム参照配列のみを用いることでは不十分であろう.今後,より多くの日本人の全ゲノム配列を決定することにより,日本人のための研究の基盤づくりが期待されるとともに,全ゲノム配列の解析によっていろいろな疾患に関連する未知の多様性が発見されるものと考えられる.また,今回の解析技術をすでに開始しているがんゲノム解析を行う国際がんゲノムコンソーシアム(International Cancer Genome Consortium:ICGC)で活用することにより,がんゲノムの包括的な情報を解明しがんの原因にせまることができるであろう.これらの結果,オーダーメイド医療がますます進展し,医学および医療に大きな展開をもたらすことになろう.
文 献
- Ozaki, K., Ohnishi, Y., Iida, A. et al.: Functional SNPs in the lymphotoxin-α gene that are associated with susceptibility to myocardial infarction. Nat. Genet., 32, 650-654 (2002)[PubMed]
- Wellcome Trust Case Control Consortium: Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature, 447, 661-678 (2007)[PubMed]
- Li, B. & Leal, S. M.: Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am. J. Human Genet., 83, 311-321 (2008)[PubMed]
- Pritchard, J. K.: Are rare variants responsible for susceptibility to complex diseases? Am. J. Human Genet., 69, 124-137 (2001)[PubMed]
- Gorlov, I. P., Gorlova, O. Y., Sunyaev, S. R. et al.: Shifting paradigm of association studies: value of rare single-nucleotide polymorphisms. Am. J. Human Genet., 82, 100-112 (2008)[PubMed]
- Nejentsev, S., Walker, N., Riches, D. et al.: Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science, 324, 387-389 (2009)[PubMed]
- Kruglyak, L.: The road to genome-wide association studies. Nat. Rev. Genet., 9, 314-318 (2008)[PubMed]
- Frazer, K. A., Murray, S. S., Schork, N. J. et al.: Human genetic variation and its contribution to complex traits. Nat. Rev. Genet., 10, 241-251 (2009)[PubMed]
- Tucker, T., Marra, M. & Friedman, J. M.: Massively parallel sequencing: the next big thing in genetic medicine. Am. J. Human Genet., 85, 142-154 (2009)[PubMed]
- The International HapMap Consorthium: A haplotype map of the human genome. Nature, 437, 1299-1320 (2005)[PubMed]
著者プロフィール
略歴:1995年 東京大学大学院工学系研究科博士課程 修了,同年 京都大学大学院工学研究科 助手,1997年 東京大学医科学研究所 リサーチアソシエイト,1998年 同 助手を経て,2000年より理化学研究所遺伝子多型研究センター(現 ゲノム医科学研究センター)チームリーダー.医学博士・工学博士.
研究テーマ:ゲノム医科学,遺伝統計学など.全ゲノムの配列の多様性や遺伝子発現の個人差と疾患との関係を解明する.
関心事:これからの日本のゲノム医科学には,大局的な視野からの計画,実験技術の進展と,情報科学や数学と医学との同時遂行,そして,多くの患者さんや病院,コーディネーターの方々の多大なご協力が必須だと思っている.
© 2010 角田 達彦 Licensed under CC 表示 2.1 日本
