miR-183クラスターは転写因子Foxo1の発現を負に制御することによりTh17細胞の病原性を促進する
市山健司1・Chen Dong 2
(1米国Children’s National Medical Center,Center for Cancer and Immunology Research,2中国Tsinghua大学Institute for Immunology)
email:市山健司
DOI: 10.7875/first.author.2016.074
The microRNA-183-96-182 cluster promotes T helper 17 cell pathogenicity by negatively regulating transcription factor Foxo1 expression.
Kenji Ichiyama, Alicia Gonzalez-Martin, Byung-Seok Kim, Hyun Yong Jin, Wei Jin, Wei Xu, Mohsen Sabouri-Ghomi, Shunbin Xu, Pan Zheng, Changchun Xiao, Chen Dong
Immunity, 44, 1284-1298 (2016)
Th17細胞はインターロイキン17を高産生するヘルパーT細胞であり,自己免疫疾患や細菌の感染に対する防御などにおいて重要な役割を担う.ナイーブT細胞からTh17細胞への分化においてはさまざまなサイトカインや転写因子の関与することが報告されている.しかしながら,非コードRNAの一種であるmiRNAのTh17細胞の分化および機能における役割については大部分が不明であった.今回,筆者らは,Th17細胞において特異的にmiRNAの機能を欠損させたマウスの解析から,miRNAはTh17細胞の病原性の獲得において必須の役割を担うことを見い出した.さらに,次世代シークエンサーを用いてmiRNAの発現を網羅的に解析することにより,Th17細胞に特異的に発現するmiRNAとしてmiR-183クラスターを同定した.miR-183クラスターはインターロイキン6-STAT3シグナル伝達系を介して発現が誘導され,転写因子Foxo1の発現の抑制を介してインターロイキン1受容体1の発現を亢進することによりTh17細胞の病原性を促進し,また,マウスにおける多発性硬化症のモデルである実験的自己免疫性脳脊髄炎の病態を悪化させた.この研究は,これまであまり知られていなかったTh17細胞の病原性の獲得における新規の制御機構を示したものであり,自己免疫疾患に対する新規の治療法の開発のための基盤の確立に大きく貢献すると思われる.
免疫系における恒常性の維持においてはヘルパーT細胞が主要な役割をはたす.ヘルパーT細胞のうち,インターロイキン17を高産生するTh17細胞は関節炎リウマチ,多発性硬化症,乾癬,炎症性腸疾患などの自己免疫疾患をひき起こす1).Th17細胞はナイーブT細胞からマスター転写因子であるRORγtの発現をともない非病原性Th17細胞あるいは病原性Th17細胞へと分化する2,3).非病原性Th17細胞はインターロイキン6およびTGFβにより分化が誘導され,インターロイキン10やインターロイキン21を高産生する.一方,病原性Th17細胞はインターロイキン1β,インターロイキン6,インターロイキン23により分化が誘導され,顆粒球単球コロニー刺激因子,インターロイキン22,インターフェロンγを高産生する.このように,Th17細胞の分化は特異的なサイトカインおよび転写因子により厳密に制御されているが,miRNAがTh17細胞におよぼす影響についてはこれまでよくわかっていなかった.この研究においては,Th17細胞において特異的にmiRNAの機能を欠失したマウスを作製し,さらに,次世代シークエンサーを用いてmiRNAの発現を網羅的に解析することにより,miRNAによるTh17細胞の制御機構の解明を試みた.
miRNAはpri-miRNAとしてゲノムから転写され,DroshaおよびDicerとよばれる酵素により成熟型のmiRNAへと変換されて遺伝子発現の抑制において機能する.したがって,DroshaあるいはDicerを欠損したマウスにおいては成熟型のmiRNAが産生されず,miRNAの機能の欠失したマウスになる.Th17細胞におけるmiRNAの役割について検討するため,Th17細胞において特異的にDicerを欠損したマウスを作製した.マウスにおける多発性硬化症のモデルである実験的自己免疫性脳脊髄炎の病態に対し,Th17細胞は深く関与する4).そこで,Th17細胞に特異的なDicerノックアウトマウスに実験的自己免疫性脳脊髄炎を発症させることにより,miRNAがTh17細胞におよぼす影響を個体のレベルにおいて検討した.その結果,対照となるマウスにおいては実験的自己免疫性脳脊髄炎が有意に発症したのに対し,Th17細胞に特異的なDicerノックアウトマウスにおいてはまったく発症せず,さらに,中枢神経系におけるTh17細胞の割合もいちじるしく低下していた.これらのことから,miRNAはTh17細胞の分化および機能において必須の役割を担うことが明らかにされた.
Th17細胞において特異的に発現するmiRNAを同定するため,in vitroにおいて分化させた各種のレポーターマウスに由来する制御性T細胞,Th1細胞,Th17細胞に対し,次世代シークエンサーを用いたRNA-seq法によりmiRNAの発現を網羅的に解析した.その結果,Th17細胞,とくに病原性Th17細胞において特異的に発現の高いmiRNAとして,miR-183,miR-96,miR-182からなるmiR-183クラスターが同定された.miR-183クラスターは感覚器官に特異的なmiRNAのクラスターとして同定され,網膜の正常な発達や機能に必須の役割を担うことが知られている5).しかしながら,免疫細胞,とくにTh17細胞においてその役割は不明であった.そこで,miR-183クラスターの発現が病原性Th17細胞においてどのように誘導されるのかを検討するため,病原性Th17細胞の分化に必要なサイトカインであるインターロイキン1β,インターロイキン6,インターロイキン23によりそれぞれT細胞を刺激し,miR-183クラスターが発現するかどうかを定量PCR法により検討した.その結果,インターロイキン6による刺激のみでmiR-183クラスターの発現は顕著に誘導された.さらに,インターロイキン6の下流の主要な転写因子であるSTAT3を欠損したT細胞においては,インターロイキン6によるmiR-183クラスターの発現の誘導は完全に消失した.これらのことから,病原性Th17細胞におけるmiR-183Cクラスターの発現はインターロイキン6-STAT3シグナル伝達系を介して誘導されることが明らかにされた.
miR-183クラスターが病原性Th17細胞におよぼす影響について検討するため,レトロウイルスの系を用いてT細胞においてmiR-183クラスターを過剰に発現させた.その結果,病原性Th17細胞に特徴的な遺伝子の発現が亢進し,病原性Th17細胞の分化が顕著に促進された.また,逆に,miR-183クラスターを欠損したマウスを作製したところ,病原性Th17細胞に特徴的な遺伝子の発現が低下し,病原性Th17細胞の分化は有意に抑制された.さらに,個体のレベルにおける影響について検討するため,miR-183クラスターノックアウトマウスに由来するCD4陽性T細胞を免疫不全マウスに移入したのち実験的自己免疫性脳脊髄炎を発症させたところ,対照となるマウスと比較して病態は有意に減弱した.これらのことから,miR-183クラスターはTh17細胞の病原性を促進することが明らかにされた.
miRNAは標的となる遺伝子の3’側非翻訳領域と結合し,翻訳の抑制やmRNAの分解を介してその遺伝子の発現を抑制する6).miR-183クラスターの標的となる遺伝子を同定するため,TargetScan algorithm 7) およびmiRDB algorithm 8) を用いてin silicoにおいて解析した.その結果,転写因子Foxo1をコードするFoxo1遺伝子はその3’側非翻訳領域にmiR-183クラスターの結合配列をもち,この配列は進化的に高度に保存されていた.さらに,in vitroにおける解析において,miR-183クラスターを過剰に発現させたTh17細胞においてFoxo1の発現が抑制され,逆に,miR-183クラスターを欠損したTh17細胞においてはFoxo1の発現が亢進したことから,実際に,Th17細胞においてFoxo1遺伝子がmiR-183クラスターの標的遺伝子である可能性が示唆された.
miR-183クラスターがFoxo1遺伝子の3’側非翻訳領域に直接に作用してその発現を制御するのかどうか検討するため,蛍光タンパク質RFPをコードする遺伝子の3’側にFoxo1遺伝子の野生型の3’側非翻訳領域あるいはmiR-183クラスターの結合配列を欠損させた3’側非翻訳領域を導入したレポーター遺伝子を作製し,RFPの発現をフローサイトメトリーを用いて測定した.その結果,miR-183クラスターの過剰な発現により,野生型の3’側非翻訳領域を導入したレポーター遺伝子の発現は減弱したが,miR-183クラスターの結合配列を欠損させた3’側非翻訳領域を導入したレポーター遺伝子の発現は減弱しなかった.したがって,miR-183クラスターはFoxo1遺伝子の3’側非翻訳領域に存在するmiR-183クラスターの結合配列と直接に結合してFoxo1の発現を抑制することが示唆された.以上の結果から,転写因子Foxo1をコードするFoxo1遺伝子はTh17細胞においてmiR-183クラスターの標的遺伝子であることが明らかにされた.
転写因子Foxo1がTh17細胞におよぼす影響について検討するため,レトロウイルスの系を用いてFoxo1を過剰に発現させた.その結果,病原性Th17細胞の分化は顕著に抑制された.また逆に,Th17細胞において特異的にFoxo1を欠損したマウスを作製し,Th17細胞の分化におよぼす影響について検討したところ,病原性Th17細胞に特徴的な遺伝子の発現が亢進し,病原性Th17細胞の分化が顕著に促進された.さらに,個体のレベルにおける影響について検討するため,Th17細胞に特異的なFoxo1ノックアウトマウスに実験的自己免疫性脳脊髄炎を発症させたところ,対照となるマウスと比較して病態は有意に悪化した.これらのことから,Foxo1はTh17細胞の病原性を抑制することが示唆された.
miR-183クラスターによるTh17細胞の病原性の促進にFoxo1が関与しているかどうか検討するため,miR-183クラスターを欠損したTh17細胞においてshRNAを用いてFoxo1をノックダウンした.その結果,miR-183クラスターの欠損にともなう病原性Th17細胞の分化の減少は野生型のマウスに由来する病原性Th17細胞と同じ程度まで回復した.以上の結果から,miR-183クラスターはFoxo1の発現の抑制を介してTh17細胞の病原性を促進することが明らかにされた.
これまでに,インターロイキン1はTh17細胞を介した自己免疫疾患の発症に重要な役割を担うこと,および,病原性Th17細胞はインターロイキン1受容体1を高発現することが知られている9,10).そこで,Foxo1によるTh17細胞の病原性の抑制にインターロイキン1受容体1が関与するかどうか検討した.インターロイキン1受容体1の発現を定量PCR法により確認したところ,Foxo1を過剰に発現したTh17細胞においてはインターロイキン1受容体1の発現は低下し,一方,Foxo1を欠損したTh17細胞においては上昇していた.Foxo1によるインターロイキン1受容体1の発現の抑制について詳細な分子機構を明らかにするため,Foxo1がインターロイキン1受容体1をコードする遺伝子のプロモーターおよびエンハンサーにおよぼす影響についてルシフェラーゼアッセイおよびクロマチン免疫沈降法により検討した.その結果,Foxo1がインターロイキン1受容体1をコードする遺伝子のエンハンサーと結合し,RORγtのエンハンサーへのリクルートを阻害することにより,その転写を強く抑制することが見い出された.
miR-183クラスターによるTh17細胞の病原性の促進にインターロイキン1受容体1が関与しているかどうか検討するため,レトロウイルスの系を用いてmiR-183クラスターを欠損したTh17細胞にインターロイキン1受容体1を過剰に発現させた.その結果,miR-183クラスターの欠損にともなう病原性Th17細胞の分化の減少は対照となるマウスと同じ程度にまで回復した.さらに,個体のレベルにおける影響について検討するため,Th17細胞を免疫不全マウスに移入したのち実験的自己免疫性脳脊髄炎を発症させた.野生型のマウスに由来するTh17細胞の移入と比較して,miR-183クラスターを欠損したTh17細胞を移入したときには病態は減弱したのに対し,インターロイキン1受容体1を過剰に発現させたmiR-183クラスターを欠損したTh17細胞を移入したときには病態の減弱はみられなかった.以上の結果から,miR-183クラスターによるFoxo1を介したインターロイキン1受容体1の発現の制御がTh17細胞の病原性を促進することが明らかにされた.
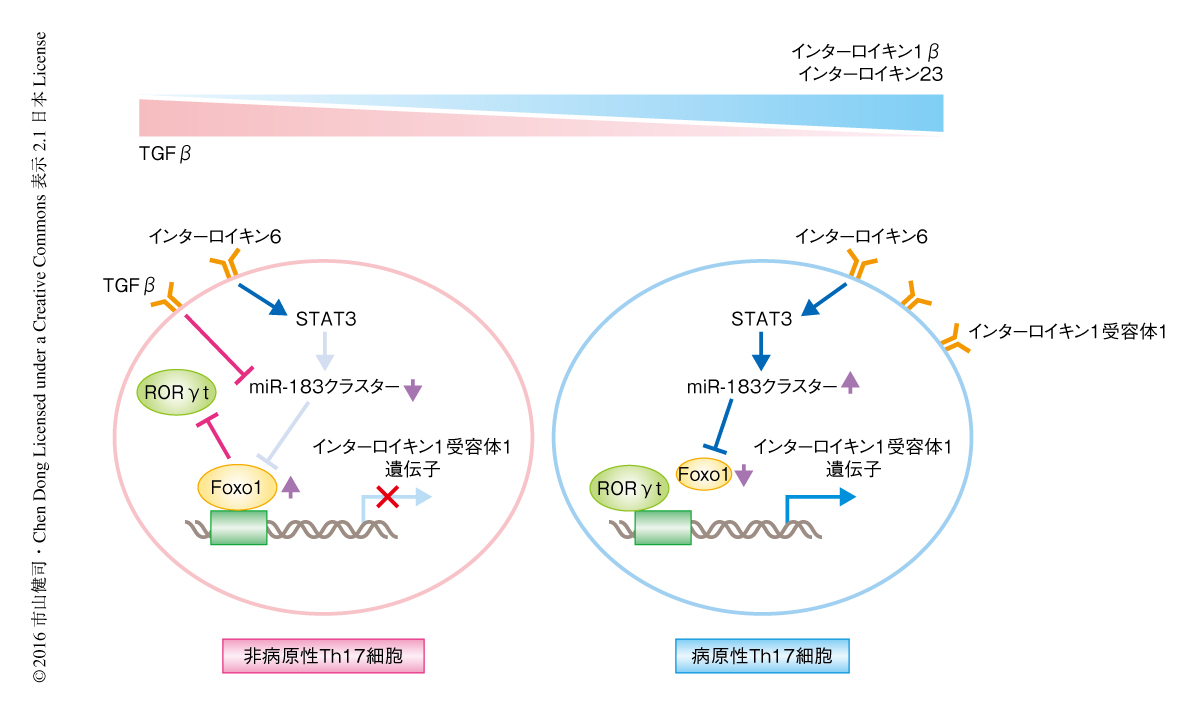
この研究により,インターロイキン6-STAT3シグナル伝達系の活性化→miR-183クラスターの発現→転写因子Foxo1の発現の抑制→インターロイキン1受容体1の発現の亢進→Th17細胞の病原性の促進,というmiRNAによるTh17細胞の新規の制御機構が明らかにされた(図1).このことから,miR-183クラスターやFoxo1の発現あるいは機能を制御することにより,Th17細胞を介して,さまざまな自己免疫疾患の制御が可能になると考えられる.しかしながら,miRNAは多様な標的遺伝子を介してその作用を示すことが知られており,今回,明らかにされた経路はmiR-183クラスターによるTh17細胞の制御機構のあくまで一部と考えられる.したがって,今後,さらなる解析によりmiR-183クラスターによるTh17細胞の病原性の制御機構の理解の深まることが,自己免疫疾患の新規の治療法の開発につながると期待される.
略歴:2009年 九州大学大学院医学研究科博士課程 修了,同年 慶應義塾大学医学部 博士研究員,2011年 米国Texas大学MD Anderson Cancer Center博士研究員,2015年 米国Children’s National Medical Centerリサーチアソシエイトを経て,2016年より大阪大学免疫学フロンティア研究センター 特任助教.
研究テーマ:非コードRNAによるヘルパーT細胞の分化の制御機構.
抱負:T細胞の研究を難治免疫疾患の新規の治療法の開発につなげたい.
Chen Dong
中国Tsinghua大学 教授.
© 2016 市山健司・Chen Dong Licensed under CC 表示 2.1 日本
(1米国Children’s National Medical Center,Center for Cancer and Immunology Research,2中国Tsinghua大学Institute for Immunology)
email:市山健司
DOI: 10.7875/first.author.2016.074
The microRNA-183-96-182 cluster promotes T helper 17 cell pathogenicity by negatively regulating transcription factor Foxo1 expression.
Kenji Ichiyama, Alicia Gonzalez-Martin, Byung-Seok Kim, Hyun Yong Jin, Wei Jin, Wei Xu, Mohsen Sabouri-Ghomi, Shunbin Xu, Pan Zheng, Changchun Xiao, Chen Dong
Immunity, 44, 1284-1298 (2016)
要 約
Th17細胞はインターロイキン17を高産生するヘルパーT細胞であり,自己免疫疾患や細菌の感染に対する防御などにおいて重要な役割を担う.ナイーブT細胞からTh17細胞への分化においてはさまざまなサイトカインや転写因子の関与することが報告されている.しかしながら,非コードRNAの一種であるmiRNAのTh17細胞の分化および機能における役割については大部分が不明であった.今回,筆者らは,Th17細胞において特異的にmiRNAの機能を欠損させたマウスの解析から,miRNAはTh17細胞の病原性の獲得において必須の役割を担うことを見い出した.さらに,次世代シークエンサーを用いてmiRNAの発現を網羅的に解析することにより,Th17細胞に特異的に発現するmiRNAとしてmiR-183クラスターを同定した.miR-183クラスターはインターロイキン6-STAT3シグナル伝達系を介して発現が誘導され,転写因子Foxo1の発現の抑制を介してインターロイキン1受容体1の発現を亢進することによりTh17細胞の病原性を促進し,また,マウスにおける多発性硬化症のモデルである実験的自己免疫性脳脊髄炎の病態を悪化させた.この研究は,これまであまり知られていなかったTh17細胞の病原性の獲得における新規の制御機構を示したものであり,自己免疫疾患に対する新規の治療法の開発のための基盤の確立に大きく貢献すると思われる.
はじめに
免疫系における恒常性の維持においてはヘルパーT細胞が主要な役割をはたす.ヘルパーT細胞のうち,インターロイキン17を高産生するTh17細胞は関節炎リウマチ,多発性硬化症,乾癬,炎症性腸疾患などの自己免疫疾患をひき起こす1).Th17細胞はナイーブT細胞からマスター転写因子であるRORγtの発現をともない非病原性Th17細胞あるいは病原性Th17細胞へと分化する2,3).非病原性Th17細胞はインターロイキン6およびTGFβにより分化が誘導され,インターロイキン10やインターロイキン21を高産生する.一方,病原性Th17細胞はインターロイキン1β,インターロイキン6,インターロイキン23により分化が誘導され,顆粒球単球コロニー刺激因子,インターロイキン22,インターフェロンγを高産生する.このように,Th17細胞の分化は特異的なサイトカインおよび転写因子により厳密に制御されているが,miRNAがTh17細胞におよぼす影響についてはこれまでよくわかっていなかった.この研究においては,Th17細胞において特異的にmiRNAの機能を欠失したマウスを作製し,さらに,次世代シークエンサーを用いてmiRNAの発現を網羅的に解析することにより,miRNAによるTh17細胞の制御機構の解明を試みた.
1.miRNAはTh17細胞の機能に必須の役割を担う
miRNAはpri-miRNAとしてゲノムから転写され,DroshaおよびDicerとよばれる酵素により成熟型のmiRNAへと変換されて遺伝子発現の抑制において機能する.したがって,DroshaあるいはDicerを欠損したマウスにおいては成熟型のmiRNAが産生されず,miRNAの機能の欠失したマウスになる.Th17細胞におけるmiRNAの役割について検討するため,Th17細胞において特異的にDicerを欠損したマウスを作製した.マウスにおける多発性硬化症のモデルである実験的自己免疫性脳脊髄炎の病態に対し,Th17細胞は深く関与する4).そこで,Th17細胞に特異的なDicerノックアウトマウスに実験的自己免疫性脳脊髄炎を発症させることにより,miRNAがTh17細胞におよぼす影響を個体のレベルにおいて検討した.その結果,対照となるマウスにおいては実験的自己免疫性脳脊髄炎が有意に発症したのに対し,Th17細胞に特異的なDicerノックアウトマウスにおいてはまったく発症せず,さらに,中枢神経系におけるTh17細胞の割合もいちじるしく低下していた.これらのことから,miRNAはTh17細胞の分化および機能において必須の役割を担うことが明らかにされた.
2.miR-183クラスターは病原性Th17細胞においてインターロイキン6-STAT3シグナル伝達系を介して特異的に発現する
Th17細胞において特異的に発現するmiRNAを同定するため,in vitroにおいて分化させた各種のレポーターマウスに由来する制御性T細胞,Th1細胞,Th17細胞に対し,次世代シークエンサーを用いたRNA-seq法によりmiRNAの発現を網羅的に解析した.その結果,Th17細胞,とくに病原性Th17細胞において特異的に発現の高いmiRNAとして,miR-183,miR-96,miR-182からなるmiR-183クラスターが同定された.miR-183クラスターは感覚器官に特異的なmiRNAのクラスターとして同定され,網膜の正常な発達や機能に必須の役割を担うことが知られている5).しかしながら,免疫細胞,とくにTh17細胞においてその役割は不明であった.そこで,miR-183クラスターの発現が病原性Th17細胞においてどのように誘導されるのかを検討するため,病原性Th17細胞の分化に必要なサイトカインであるインターロイキン1β,インターロイキン6,インターロイキン23によりそれぞれT細胞を刺激し,miR-183クラスターが発現するかどうかを定量PCR法により検討した.その結果,インターロイキン6による刺激のみでmiR-183クラスターの発現は顕著に誘導された.さらに,インターロイキン6の下流の主要な転写因子であるSTAT3を欠損したT細胞においては,インターロイキン6によるmiR-183クラスターの発現の誘導は完全に消失した.これらのことから,病原性Th17細胞におけるmiR-183Cクラスターの発現はインターロイキン6-STAT3シグナル伝達系を介して誘導されることが明らかにされた.
3.miR-183クラスターはTh17細胞の病原性を促進する
miR-183クラスターが病原性Th17細胞におよぼす影響について検討するため,レトロウイルスの系を用いてT細胞においてmiR-183クラスターを過剰に発現させた.その結果,病原性Th17細胞に特徴的な遺伝子の発現が亢進し,病原性Th17細胞の分化が顕著に促進された.また,逆に,miR-183クラスターを欠損したマウスを作製したところ,病原性Th17細胞に特徴的な遺伝子の発現が低下し,病原性Th17細胞の分化は有意に抑制された.さらに,個体のレベルにおける影響について検討するため,miR-183クラスターノックアウトマウスに由来するCD4陽性T細胞を免疫不全マウスに移入したのち実験的自己免疫性脳脊髄炎を発症させたところ,対照となるマウスと比較して病態は有意に減弱した.これらのことから,miR-183クラスターはTh17細胞の病原性を促進することが明らかにされた.
4.Foxo1をコードする遺伝子はTh17細胞においてmiR-183クラスターの標的となる
miRNAは標的となる遺伝子の3’側非翻訳領域と結合し,翻訳の抑制やmRNAの分解を介してその遺伝子の発現を抑制する6).miR-183クラスターの標的となる遺伝子を同定するため,TargetScan algorithm 7) およびmiRDB algorithm 8) を用いてin silicoにおいて解析した.その結果,転写因子Foxo1をコードするFoxo1遺伝子はその3’側非翻訳領域にmiR-183クラスターの結合配列をもち,この配列は進化的に高度に保存されていた.さらに,in vitroにおける解析において,miR-183クラスターを過剰に発現させたTh17細胞においてFoxo1の発現が抑制され,逆に,miR-183クラスターを欠損したTh17細胞においてはFoxo1の発現が亢進したことから,実際に,Th17細胞においてFoxo1遺伝子がmiR-183クラスターの標的遺伝子である可能性が示唆された.
miR-183クラスターがFoxo1遺伝子の3’側非翻訳領域に直接に作用してその発現を制御するのかどうか検討するため,蛍光タンパク質RFPをコードする遺伝子の3’側にFoxo1遺伝子の野生型の3’側非翻訳領域あるいはmiR-183クラスターの結合配列を欠損させた3’側非翻訳領域を導入したレポーター遺伝子を作製し,RFPの発現をフローサイトメトリーを用いて測定した.その結果,miR-183クラスターの過剰な発現により,野生型の3’側非翻訳領域を導入したレポーター遺伝子の発現は減弱したが,miR-183クラスターの結合配列を欠損させた3’側非翻訳領域を導入したレポーター遺伝子の発現は減弱しなかった.したがって,miR-183クラスターはFoxo1遺伝子の3’側非翻訳領域に存在するmiR-183クラスターの結合配列と直接に結合してFoxo1の発現を抑制することが示唆された.以上の結果から,転写因子Foxo1をコードするFoxo1遺伝子はTh17細胞においてmiR-183クラスターの標的遺伝子であることが明らかにされた.
5.miR-183クラスターはFoxo1の発現の抑制を介してTh17細胞の病原性を促進する
転写因子Foxo1がTh17細胞におよぼす影響について検討するため,レトロウイルスの系を用いてFoxo1を過剰に発現させた.その結果,病原性Th17細胞の分化は顕著に抑制された.また逆に,Th17細胞において特異的にFoxo1を欠損したマウスを作製し,Th17細胞の分化におよぼす影響について検討したところ,病原性Th17細胞に特徴的な遺伝子の発現が亢進し,病原性Th17細胞の分化が顕著に促進された.さらに,個体のレベルにおける影響について検討するため,Th17細胞に特異的なFoxo1ノックアウトマウスに実験的自己免疫性脳脊髄炎を発症させたところ,対照となるマウスと比較して病態は有意に悪化した.これらのことから,Foxo1はTh17細胞の病原性を抑制することが示唆された.
miR-183クラスターによるTh17細胞の病原性の促進にFoxo1が関与しているかどうか検討するため,miR-183クラスターを欠損したTh17細胞においてshRNAを用いてFoxo1をノックダウンした.その結果,miR-183クラスターの欠損にともなう病原性Th17細胞の分化の減少は野生型のマウスに由来する病原性Th17細胞と同じ程度まで回復した.以上の結果から,miR-183クラスターはFoxo1の発現の抑制を介してTh17細胞の病原性を促進することが明らかにされた.
6.miR-183クラスターによるFoxo1を介したインターロイキン1受容体1の発現の制御はTh17細胞の病原性を促進する
これまでに,インターロイキン1はTh17細胞を介した自己免疫疾患の発症に重要な役割を担うこと,および,病原性Th17細胞はインターロイキン1受容体1を高発現することが知られている9,10).そこで,Foxo1によるTh17細胞の病原性の抑制にインターロイキン1受容体1が関与するかどうか検討した.インターロイキン1受容体1の発現を定量PCR法により確認したところ,Foxo1を過剰に発現したTh17細胞においてはインターロイキン1受容体1の発現は低下し,一方,Foxo1を欠損したTh17細胞においては上昇していた.Foxo1によるインターロイキン1受容体1の発現の抑制について詳細な分子機構を明らかにするため,Foxo1がインターロイキン1受容体1をコードする遺伝子のプロモーターおよびエンハンサーにおよぼす影響についてルシフェラーゼアッセイおよびクロマチン免疫沈降法により検討した.その結果,Foxo1がインターロイキン1受容体1をコードする遺伝子のエンハンサーと結合し,RORγtのエンハンサーへのリクルートを阻害することにより,その転写を強く抑制することが見い出された.
miR-183クラスターによるTh17細胞の病原性の促進にインターロイキン1受容体1が関与しているかどうか検討するため,レトロウイルスの系を用いてmiR-183クラスターを欠損したTh17細胞にインターロイキン1受容体1を過剰に発現させた.その結果,miR-183クラスターの欠損にともなう病原性Th17細胞の分化の減少は対照となるマウスと同じ程度にまで回復した.さらに,個体のレベルにおける影響について検討するため,Th17細胞を免疫不全マウスに移入したのち実験的自己免疫性脳脊髄炎を発症させた.野生型のマウスに由来するTh17細胞の移入と比較して,miR-183クラスターを欠損したTh17細胞を移入したときには病態は減弱したのに対し,インターロイキン1受容体1を過剰に発現させたmiR-183クラスターを欠損したTh17細胞を移入したときには病態の減弱はみられなかった.以上の結果から,miR-183クラスターによるFoxo1を介したインターロイキン1受容体1の発現の制御がTh17細胞の病原性を促進することが明らかにされた.
おわりに
この研究により,インターロイキン6-STAT3シグナル伝達系の活性化→miR-183クラスターの発現→転写因子Foxo1の発現の抑制→インターロイキン1受容体1の発現の亢進→Th17細胞の病原性の促進,というmiRNAによるTh17細胞の新規の制御機構が明らかにされた(図1).このことから,miR-183クラスターやFoxo1の発現あるいは機能を制御することにより,Th17細胞を介して,さまざまな自己免疫疾患の制御が可能になると考えられる.しかしながら,miRNAは多様な標的遺伝子を介してその作用を示すことが知られており,今回,明らかにされた経路はmiR-183クラスターによるTh17細胞の制御機構のあくまで一部と考えられる.したがって,今後,さらなる解析によりmiR-183クラスターによるTh17細胞の病原性の制御機構の理解の深まることが,自己免疫疾患の新規の治療法の開発につながると期待される.
文 献
- Korn, T., Bettelli, E., Oukka, M. et al.: IL-17 and Th17 cells. Annu. Rev. Immunol., 27, 485-517 (2009)[PubMed]
- Peters, A., Lee, Y. & Kuchroo, V. K.: The many faces of Th17 cells. Curr. Opin. Immunol., 23, 702-706 (2011)[PubMed]
- Ivanov, I. I., McKenzie, B. S., Zhou, L. et al.: The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell, 126, 1121-1133 (2006)[PubMed]
- Baxter, A. G.: The origin and application of experimental autoimmune encephalomyelitis. Nat. Rev. Immunol., 7, 904-912 (2007)[PubMed]
- Xu, S., Witmer, P. D., Lumayag, S. et al.: MicroRNA (miRNA) transcriptome of mouse retina and identification of a sensory organ-specific miRNA cluster. J. Biol. Chem., 282, 25053-25066 (2007)[PubMed]
- Bartel, D. P.: MicroRNAs: target recognition and regulatory functions. Cell, 136, 215-233 (2009)[PubMed]
- Lewis, B. P., Shih, I. H., Jones-Rhoades, M. W. et al.: Prediction of mammalian microRNA targets. Cell, 115, 787-798 (2003)[PubMed]
- Wang, X.: miRDB: a microRNA target prediction and functional annotation database with a wiki interface. RNA, 14, 1012-1017 (2008)[PubMed]
- Sutton, C., Brereton, C., Keogh, B. et al.: A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J. Exp. Med., 203, 1685-1691 (2006)[PubMed]
- Lee, Y., Awasthi, A., Yosef, N. et al.: Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol., 13, 991-999 (2012)[PubMed]
活用したデータベースにかかわるキーワードと統合TVへのリンク
著者プロフィール
略歴:2009年 九州大学大学院医学研究科博士課程 修了,同年 慶應義塾大学医学部 博士研究員,2011年 米国Texas大学MD Anderson Cancer Center博士研究員,2015年 米国Children’s National Medical Centerリサーチアソシエイトを経て,2016年より大阪大学免疫学フロンティア研究センター 特任助教.
研究テーマ:非コードRNAによるヘルパーT細胞の分化の制御機構.
抱負:T細胞の研究を難治免疫疾患の新規の治療法の開発につなげたい.
Chen Dong
中国Tsinghua大学 教授.
© 2016 市山健司・Chen Dong Licensed under CC 表示 2.1 日本
