心筋のサルコメアの収縮に対する低分子阻害剤によるマウスにおける肥大型心筋症の抑制
脇本 博子
(米国Harvard Medical School,Department of Genetics)
email:脇本博子
DOI: 10.7875/first.author.2016.013
A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice.
Eric M. Green, Hiroko Wakimoto, Robert L. Anderson, Marc J. Evanchik, Joshua M. Gorham, Brooke C. Harrison, Marcus Henze, Raja Kawas, Johan D. Oslob, Hector M. Rodriguez, Yonghong Song, William Wan, Leslie A. Leinwand, James A. Spudich, Robert S. McDowell, J. G. Seidman, Christine E. Seidman
Science, 351, 617-621 (2016)
肥大型心筋症は心筋のサルコメアを構成するタンパク質の変異などに起因する遺伝性の疾患である.肥大型心筋症は心筋細胞の肥大を特徴とするが,初期には心筋の過剰な収縮および拡張の障害などの異常が認められる.除膜した筋原線維,心筋細胞,組換えタンパク質などを用いた生体外における研究においては変異をもつ心筋は収縮力が亢進あるいは減弱するとの相反する結果が報告され,現在も争点になっている.筆者らは,心筋細胞のミオシンのもつATP分解酵素活性に対する低分子阻害剤としてMYK-461を同定し,さらに,MYK-461が心筋のサルコメアの収縮を抑制する作用をもつことを確認した.MYK-461の投与はミオシンに変異をもつ肥大型心筋症のモデルマウスにおいて心室壁の肥厚,心筋における錯綜配列の形成,心筋の線維化を抑制した.また,MYK-461を投与した肥大型心筋症のモデルマウスにおいては心筋細胞の肥大および心筋の線維化に関連する遺伝子発現の変化も認められた.この研究により,生体内における心筋のサルコメアの収縮を直接的に抑制する低分子阻害剤の投与により肥大型心筋症が改善することが示され,このことは,心筋のサルコメアの過剰な収縮が肥大型心筋症の病態の本質であり,これを抑制することが肥大型心筋症の治療につながることを示唆した.
肥大型心筋症は遺伝性の心筋の疾患としてはもっとも高頻度に認められ,米国では成人500人に1人の割合で発症する1).その病態はほかに原因のない心室壁の肥厚であり,特徴的な病理所見として,心筋細胞の肥大,心筋における錯綜配列の形成,心筋の線維化があげられる.肥大型心筋症には心筋の過剰な収縮および拡張の障害など血行の動態に異常が認められ,運動誘発性の狭心症,不整脈,心不全などを発症する致死性の疾患である.現在のところ有効な根治的な治療法はなく,βアドレナリン阻害剤やCa2+チャネル阻害剤などの投与は症状の軽減に有効とされるが,疾患の進行を抑えることはできない.肥大型心筋症を発症する遺伝子変異は心筋細胞のミオシン重鎖をコードするMYH7遺伝子およびミオシン結合タンパク質をコードするMYBPC3遺伝子においてもっとも多く認められる.MYH7遺伝子あるいはMYBPC3遺伝子に変異のある場合,心室壁の肥厚が発症するまえに心筋の過剰な収縮および拡張の障害が認められる2-4).
近年,これらの遺伝子変異のもたらす生化学的および生理学的な変化を分子レベルで明らかにし,これが肥大型心筋症の患者に認められる臨床症状といかに関連するかを明らかにする研究がさかんである.肥大型心筋症のモデルマウスから単離した心筋細胞,ヒトの筋原線維の標本,変異型の組換えミオシンなどを使ったさまざまな研究において,ミオシンのもつATP分解酵素活性の亢進,筋緊張の亢進,アクチンフィラメントの滑り速度の上昇などが報告されており5,6),これらは肥大型心筋症の患者に認められる心筋の過剰な収縮との関連が強く示唆されている.心臓の線維芽細胞は心筋細胞からの機械的な刺激,分泌物,電気生理学的なシグナルに反応し,心筋の線維化を誘発する遺伝子,あるいは,細胞外マトリックスを構成するタンパク質の産生を促進する遺伝子の発現を亢進することが知られている.異常をもつミオシンによる過剰な機械的な刺激によりこれらの遺伝子発現が上昇し,肥大型心筋症にみられる心筋の線維化などの病理像を発症するという説は合理的と考えられる.しかしながら,組換えミオシンを用いた研究において,変異型のミオシンは心筋のサルコメアの仕事率を上昇させず,わずかながら力の発生を減少させると報告するものもあり7),肥大型心筋症の原因となる遺伝子変異が分子レベルにおいて心筋のサルコメアの仕事率を上昇させるかどうかについてはいまだ結論がでていない.
組換えタンパク質あるいは筋原線維の標本を用いた力の発生や滑り速度の測定など生体外における実験系には限界があるため,肥大型心筋症のモデルマウスに心筋のサルコメアの仕事率を低下させるような低分子阻害剤を投与し,心筋のサルコメアの収縮を抑制させるという生体内における実験を試みた.つまり,心筋のサルコメアの仕事率の異常な上昇が肥大型心筋症の本態であるならば,これに対する低分子阻害剤を生体に投与することにより肥大型心筋症の発症は抑制され特徴的な病理像は改善されるとの仮説をたてた.心筋のサルコメアの仕事率はミオシンの発生する力とミオシンとアクチンとの滑り速度により決定されるので,理論的には,力あるいは滑り速度のいずれかを抑えることにより仕事率は低下する.そこで,両者のうち力の発生を抑えることを目的とし,ウシの筋原線維を用いた化学的なスクリーニングにてアクチンにより活性化されるミオシンのATP分解酵素活性の最大活性を低下させる分子を探索しMYK-461を同定した(図1).
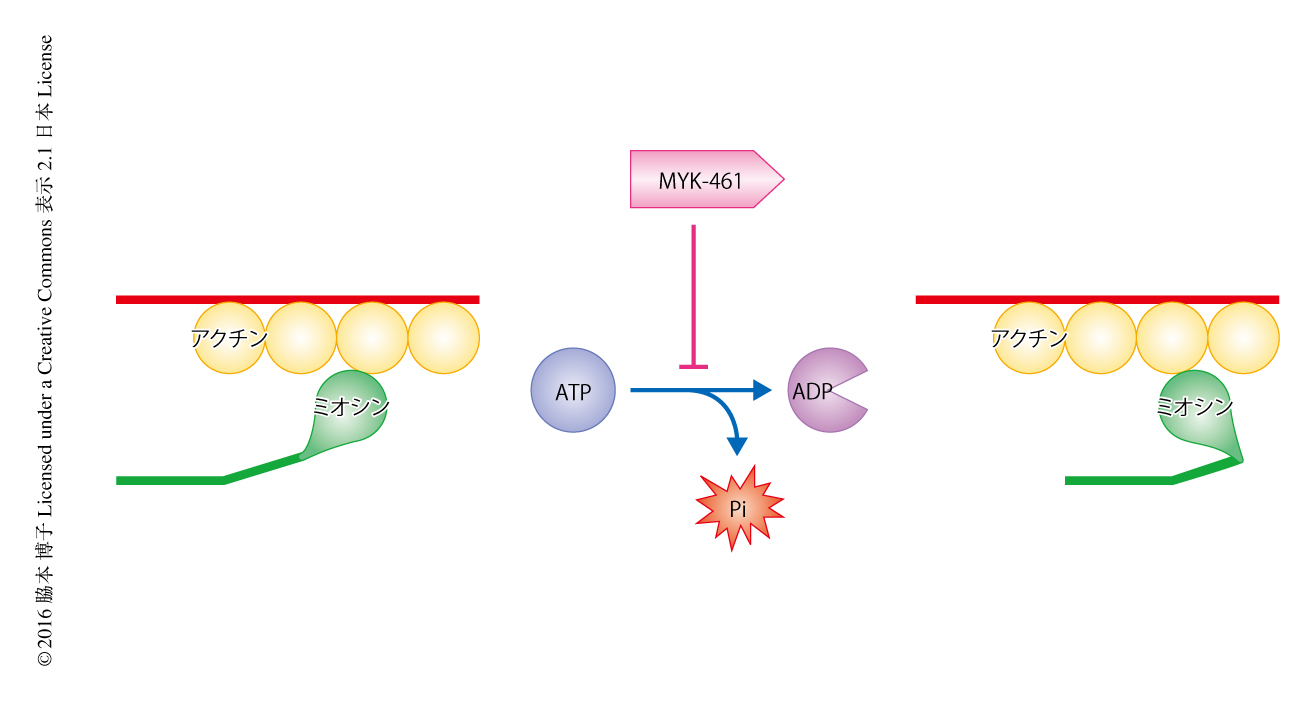
マウスの心筋の筋原線維においてATP分解酵素活性を測定したところ,MYK-461は用量に依存して活性を抑制した.また,ラットの除膜した心筋線維においてMYK-461は細胞内Ca2+濃度にかかわらず用量に依存して最大張力を減少させた.さらに,ラットから単離した心筋細胞においてMYK-461は細胞内Ca2+濃度にかかわらず用量に依存して心筋細胞の収縮率を低下させた.
筆者らは,これまで,肥大型心筋症の患者のMYH7遺伝子にて同定されたミスセンス変異をマウスに導入することにより複数の肥大型心筋症のモデルマウスを作製してきた.これらのモデルマウスは形態および血行の動態などにおいてヒトの肥大型心筋症をよく模しており,これらを用いて肥大型心筋症の発症の機序およびその治療法の開発につき研究してきた.今回,生体におけるMYK-461の効果を調べるため,これらのマウスモデルのうち,アクチン結合部位,核酸結合部位に隣接した部位,コンバータードメインにそれぞれ,1アミノ酸置換をヘテロ接合体としてもつ3つの系統を利用した.MYK-461は飲料水に混ぜ経口にて投与した.6~15週齢の若年のマウスに対し投与を開始したのち,2~4週ごとに心機能の検査および血中のMYK-461の濃度の測定を施行したところ,野生型のマウスおよび肥大型心筋症モデルマウスとも,MYK-461の投与により心臓の収縮率は低下し,血中のMYK-461の濃度と心臓の収縮率とは負の線形相関を示した.一方で,握力の測定などにおいて筋力の低下など骨格筋に対する明らかな影響は認められなかった.
心筋症を発症するまえの若年の肥大型心筋症モデルマウスおよび対照となる野生型のマウスに対し長期にわたりMYK-461を投与したところ,MYK-461を投与していない肥大型心筋症モデルマウスにおいては心臓の肥大が進行性に発症したのに対し,MYK-461を投与した肥大型心筋症モデルマウスの心室壁の厚さは野生型マウスと有意な差がなかった.すでに心筋症を発症した高齢の肥大型心筋症モデルマウスにMYK-461を投与したところ,2週のちに心臓の収縮率の低下,4週のちに心筋壁の厚さの減少が認められた.組織学的な検査においては,MYK-461を投与していない肥大型心筋症モデルマウスにおいては心室の約5~10%の心筋に線維化が認められたのに対し,心筋症を発症するまえからMYK-461を投与した肥大型心筋症モデルマウスにおいては心筋の線維化の有意な抑制が認められた.心筋症を発症したのちにMYK-461を投与した肥大型心筋症モデルマウスにおいては心筋の線維化の有意な改善は認められなかった.肥大型心筋症の特徴的な病理所見として心筋における錯綜配列の形成があげられるが,これを定量的に評価するため,筋線維の限局的な配列の方向を決めるアルゴリズムを用いた.その結果,心筋の線維化と同様に,MYK-461を投与していない肥大型心筋症モデルマウスと比較して心筋症を発症するまえからMYK-461を投与した肥大型心筋症モデルマウスにおいては心筋における錯綜配列の形成につき改善が認められたが,肥大型心筋症を発症したのちにMYK-461を投与した肥大型心筋症モデルマウスにおいては有意な改善は認められなかった.これらの結果から,肥大型心筋症が発症するまえに心筋のサルコメアの仕事率を低下させることで,より効果的な治療結果がもたらされることが示唆された.
肥大型心筋症モデルマウスにおいて心筋のサルコメアにおける力の発生の抑制が遺伝子発現にもたらす影響について調べた.まず,心筋症を発症した肥大型心筋症モデルマウスと対照となる野生型のマウスの遺伝子発現を比較し,発現量に異常の認められた200の遺伝子を同定した.これらの遺伝子は,心筋細胞に発現し心臓の収縮に関与するタンパク質をコードするもの,および,線維芽細胞に発現し細胞外マトリックスの産生に関与するタンパク質をコードするものを多く含んでいた.心筋症を発症するまえからMYK-461を投与した肥大型心筋症モデルマウス,および,心筋症を発症したのちにMYK-461を投与した肥大型心筋症モデルマウスとも,この200の遺伝子の発現はMYK-461を投与していない肥大型心筋症モデルマウスよりも野生型のマウスに類似していた.心筋症を発症したのちにMYK-461を投与した肥大型心筋症モデルマウスにおいては心筋の線維化の正常化は認められなかったものの,肥大型心筋症を発症したのちに心筋のサルコメアにおいて力の発生を抑制した場合,心筋細胞および線維芽細胞において遺伝子発現については正常化の傾向が誘発されたと考えられた.
心室壁の肥厚はエネルギー代謝の異常にも関与することから,ミトコンドリアタンパク質をコードする1158の遺伝子の発現について調べた.その結果,MYK-461を投与していない心筋症モデルマウスにおいてはミトコンドリアタンパク質をコードする遺伝子の20~30%に発現の異常があったが,MYK-461を投与した肥大型心筋症モデルマウスにおいては約5%の異常にとどまった.このことから,肥大型心筋症は代謝の異常と関連すること,そして,MYK-461の投与はこれを正常化することが示唆された.
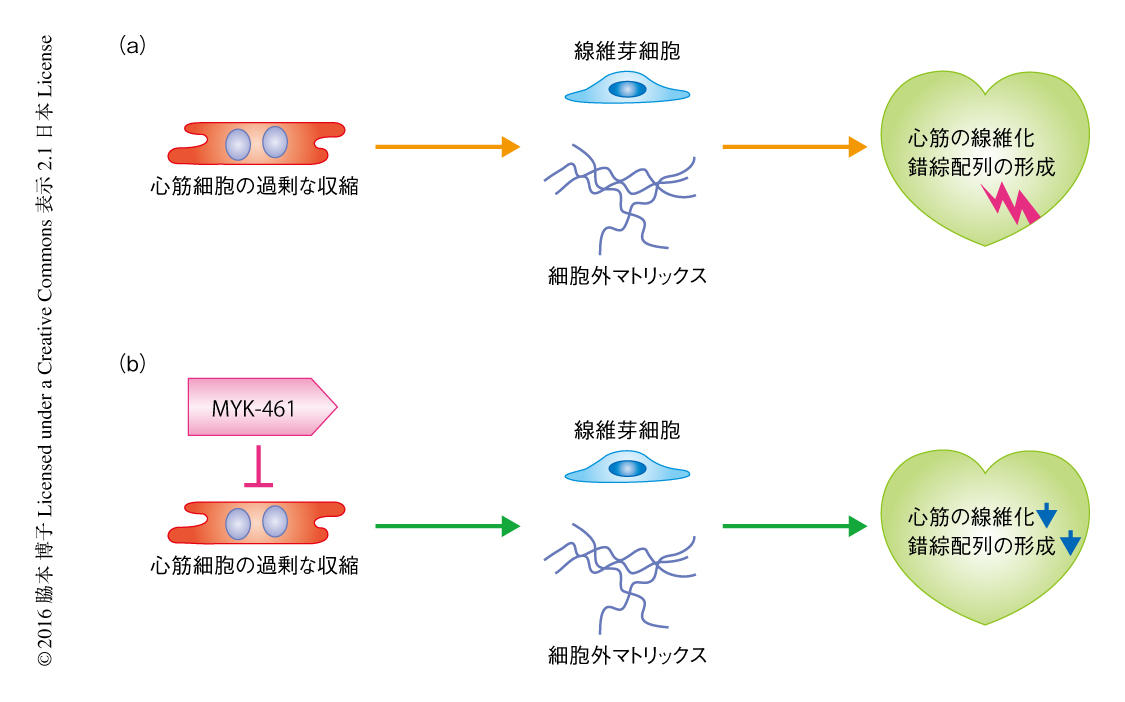
この研究において,筆者らは,心筋のサルコメアの仕事率を低下させる作用をもつ低分子阻害剤の早期からの投与が肥大型心筋症の形態学的および組織学的な異常,遺伝子発現の異常を改善することを示した.これは,肥大型心筋症の原因となる変異タンパク質が心筋のサルコメアの仕事率および収縮力を亢進し,結果として,病理的なリモデリングを促進するという説を支持すると考えられた.この結果は組換えタンパク質を用いた生体外におけるいくつかの実験の結果と相反するものであり,心筋のサルコメアを構成するあるいは仕事率の上昇に関与する重要なタンパク質が欠失している可能性のある生体外における実験系には限界のある可能性が示唆された.今回の研究は,ミオシンを直接の標的として力の発生を抑制する低分子阻害剤が生体において肥大型心筋症の病態を改善することを示し,心筋のサルコメアの仕事率を低下させることが肥大型心筋症の有力な治療法のひとつとなりうることが提唱された(図2).
拡張型心筋症あるいは心緻密化障害による心筋症など,ほかの遺伝性の心筋症においては,すでに心筋のサルコメアの仕事率の異常が報告されている8).今回,肥大型心筋症に対しても心筋のサルコメアの仕事率の正常化が病態の改善に貢献したという結果と考え合わせると,心筋のサルコメアの異常な仕事率を正常化することを目標とする薬剤の開発は心筋症の一般に共通した治療戦略として大いに期待できるものと考えられる.実際に,心不全の患者に対するミオシンの活性化薬の臨床治験はすでに開始されている.ヒトの遺伝子異常が心臓の機能にもたらす生化学的あるいは生理学的な効果につきさらに詳細に検討することにより,心筋症の根本的な発症の機序が解明され,将来的には根治療法につながることが期待される.
略歴:2000年 東京医科歯科大学大学院医歯学総合研究科にて博士号取得,米国Boston Children’s Hospital研究員,2002年 東京医科歯科大学大学院医歯学総合研究科 助手,2006年 米国Boston Children’s Hospital研究員を経て,2007年より米国Harvard Medical School講師.
研究テーマ:遺伝性および発生の異常にもとづく心臓疾患の発生の機序.
関心事:働く女性のかかえる問題点の日米での比較検討および今後の展望.
© 2016 脇本 博子 Licensed under CC 表示 2.1 日本
(米国Harvard Medical School,Department of Genetics)
email:脇本博子
DOI: 10.7875/first.author.2016.013
A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice.
Eric M. Green, Hiroko Wakimoto, Robert L. Anderson, Marc J. Evanchik, Joshua M. Gorham, Brooke C. Harrison, Marcus Henze, Raja Kawas, Johan D. Oslob, Hector M. Rodriguez, Yonghong Song, William Wan, Leslie A. Leinwand, James A. Spudich, Robert S. McDowell, J. G. Seidman, Christine E. Seidman
Science, 351, 617-621 (2016)
要 約
肥大型心筋症は心筋のサルコメアを構成するタンパク質の変異などに起因する遺伝性の疾患である.肥大型心筋症は心筋細胞の肥大を特徴とするが,初期には心筋の過剰な収縮および拡張の障害などの異常が認められる.除膜した筋原線維,心筋細胞,組換えタンパク質などを用いた生体外における研究においては変異をもつ心筋は収縮力が亢進あるいは減弱するとの相反する結果が報告され,現在も争点になっている.筆者らは,心筋細胞のミオシンのもつATP分解酵素活性に対する低分子阻害剤としてMYK-461を同定し,さらに,MYK-461が心筋のサルコメアの収縮を抑制する作用をもつことを確認した.MYK-461の投与はミオシンに変異をもつ肥大型心筋症のモデルマウスにおいて心室壁の肥厚,心筋における錯綜配列の形成,心筋の線維化を抑制した.また,MYK-461を投与した肥大型心筋症のモデルマウスにおいては心筋細胞の肥大および心筋の線維化に関連する遺伝子発現の変化も認められた.この研究により,生体内における心筋のサルコメアの収縮を直接的に抑制する低分子阻害剤の投与により肥大型心筋症が改善することが示され,このことは,心筋のサルコメアの過剰な収縮が肥大型心筋症の病態の本質であり,これを抑制することが肥大型心筋症の治療につながることを示唆した.
はじめに
肥大型心筋症は遺伝性の心筋の疾患としてはもっとも高頻度に認められ,米国では成人500人に1人の割合で発症する1).その病態はほかに原因のない心室壁の肥厚であり,特徴的な病理所見として,心筋細胞の肥大,心筋における錯綜配列の形成,心筋の線維化があげられる.肥大型心筋症には心筋の過剰な収縮および拡張の障害など血行の動態に異常が認められ,運動誘発性の狭心症,不整脈,心不全などを発症する致死性の疾患である.現在のところ有効な根治的な治療法はなく,βアドレナリン阻害剤やCa2+チャネル阻害剤などの投与は症状の軽減に有効とされるが,疾患の進行を抑えることはできない.肥大型心筋症を発症する遺伝子変異は心筋細胞のミオシン重鎖をコードするMYH7遺伝子およびミオシン結合タンパク質をコードするMYBPC3遺伝子においてもっとも多く認められる.MYH7遺伝子あるいはMYBPC3遺伝子に変異のある場合,心室壁の肥厚が発症するまえに心筋の過剰な収縮および拡張の障害が認められる2-4).
近年,これらの遺伝子変異のもたらす生化学的および生理学的な変化を分子レベルで明らかにし,これが肥大型心筋症の患者に認められる臨床症状といかに関連するかを明らかにする研究がさかんである.肥大型心筋症のモデルマウスから単離した心筋細胞,ヒトの筋原線維の標本,変異型の組換えミオシンなどを使ったさまざまな研究において,ミオシンのもつATP分解酵素活性の亢進,筋緊張の亢進,アクチンフィラメントの滑り速度の上昇などが報告されており5,6),これらは肥大型心筋症の患者に認められる心筋の過剰な収縮との関連が強く示唆されている.心臓の線維芽細胞は心筋細胞からの機械的な刺激,分泌物,電気生理学的なシグナルに反応し,心筋の線維化を誘発する遺伝子,あるいは,細胞外マトリックスを構成するタンパク質の産生を促進する遺伝子の発現を亢進することが知られている.異常をもつミオシンによる過剰な機械的な刺激によりこれらの遺伝子発現が上昇し,肥大型心筋症にみられる心筋の線維化などの病理像を発症するという説は合理的と考えられる.しかしながら,組換えミオシンを用いた研究において,変異型のミオシンは心筋のサルコメアの仕事率を上昇させず,わずかながら力の発生を減少させると報告するものもあり7),肥大型心筋症の原因となる遺伝子変異が分子レベルにおいて心筋のサルコメアの仕事率を上昇させるかどうかについてはいまだ結論がでていない.
1.ミオシンのATP分解酵素活性に対する低分子阻害剤MYK-461の同定
組換えタンパク質あるいは筋原線維の標本を用いた力の発生や滑り速度の測定など生体外における実験系には限界があるため,肥大型心筋症のモデルマウスに心筋のサルコメアの仕事率を低下させるような低分子阻害剤を投与し,心筋のサルコメアの収縮を抑制させるという生体内における実験を試みた.つまり,心筋のサルコメアの仕事率の異常な上昇が肥大型心筋症の本態であるならば,これに対する低分子阻害剤を生体に投与することにより肥大型心筋症の発症は抑制され特徴的な病理像は改善されるとの仮説をたてた.心筋のサルコメアの仕事率はミオシンの発生する力とミオシンとアクチンとの滑り速度により決定されるので,理論的には,力あるいは滑り速度のいずれかを抑えることにより仕事率は低下する.そこで,両者のうち力の発生を抑えることを目的とし,ウシの筋原線維を用いた化学的なスクリーニングにてアクチンにより活性化されるミオシンのATP分解酵素活性の最大活性を低下させる分子を探索しMYK-461を同定した(図1).
マウスの心筋の筋原線維においてATP分解酵素活性を測定したところ,MYK-461は用量に依存して活性を抑制した.また,ラットの除膜した心筋線維においてMYK-461は細胞内Ca2+濃度にかかわらず用量に依存して最大張力を減少させた.さらに,ラットから単離した心筋細胞においてMYK-461は細胞内Ca2+濃度にかかわらず用量に依存して心筋細胞の収縮率を低下させた.
2.肥大型心筋症モデルマウスに対するMYK-461の投与の効果
筆者らは,これまで,肥大型心筋症の患者のMYH7遺伝子にて同定されたミスセンス変異をマウスに導入することにより複数の肥大型心筋症のモデルマウスを作製してきた.これらのモデルマウスは形態および血行の動態などにおいてヒトの肥大型心筋症をよく模しており,これらを用いて肥大型心筋症の発症の機序およびその治療法の開発につき研究してきた.今回,生体におけるMYK-461の効果を調べるため,これらのマウスモデルのうち,アクチン結合部位,核酸結合部位に隣接した部位,コンバータードメインにそれぞれ,1アミノ酸置換をヘテロ接合体としてもつ3つの系統を利用した.MYK-461は飲料水に混ぜ経口にて投与した.6~15週齢の若年のマウスに対し投与を開始したのち,2~4週ごとに心機能の検査および血中のMYK-461の濃度の測定を施行したところ,野生型のマウスおよび肥大型心筋症モデルマウスとも,MYK-461の投与により心臓の収縮率は低下し,血中のMYK-461の濃度と心臓の収縮率とは負の線形相関を示した.一方で,握力の測定などにおいて筋力の低下など骨格筋に対する明らかな影響は認められなかった.
心筋症を発症するまえの若年の肥大型心筋症モデルマウスおよび対照となる野生型のマウスに対し長期にわたりMYK-461を投与したところ,MYK-461を投与していない肥大型心筋症モデルマウスにおいては心臓の肥大が進行性に発症したのに対し,MYK-461を投与した肥大型心筋症モデルマウスの心室壁の厚さは野生型マウスと有意な差がなかった.すでに心筋症を発症した高齢の肥大型心筋症モデルマウスにMYK-461を投与したところ,2週のちに心臓の収縮率の低下,4週のちに心筋壁の厚さの減少が認められた.組織学的な検査においては,MYK-461を投与していない肥大型心筋症モデルマウスにおいては心室の約5~10%の心筋に線維化が認められたのに対し,心筋症を発症するまえからMYK-461を投与した肥大型心筋症モデルマウスにおいては心筋の線維化の有意な抑制が認められた.心筋症を発症したのちにMYK-461を投与した肥大型心筋症モデルマウスにおいては心筋の線維化の有意な改善は認められなかった.肥大型心筋症の特徴的な病理所見として心筋における錯綜配列の形成があげられるが,これを定量的に評価するため,筋線維の限局的な配列の方向を決めるアルゴリズムを用いた.その結果,心筋の線維化と同様に,MYK-461を投与していない肥大型心筋症モデルマウスと比較して心筋症を発症するまえからMYK-461を投与した肥大型心筋症モデルマウスにおいては心筋における錯綜配列の形成につき改善が認められたが,肥大型心筋症を発症したのちにMYK-461を投与した肥大型心筋症モデルマウスにおいては有意な改善は認められなかった.これらの結果から,肥大型心筋症が発症するまえに心筋のサルコメアの仕事率を低下させることで,より効果的な治療結果がもたらされることが示唆された.
3.MYK-461を投与した肥大型心筋症のモデルマウスにおける遺伝子発現の変化
肥大型心筋症モデルマウスにおいて心筋のサルコメアにおける力の発生の抑制が遺伝子発現にもたらす影響について調べた.まず,心筋症を発症した肥大型心筋症モデルマウスと対照となる野生型のマウスの遺伝子発現を比較し,発現量に異常の認められた200の遺伝子を同定した.これらの遺伝子は,心筋細胞に発現し心臓の収縮に関与するタンパク質をコードするもの,および,線維芽細胞に発現し細胞外マトリックスの産生に関与するタンパク質をコードするものを多く含んでいた.心筋症を発症するまえからMYK-461を投与した肥大型心筋症モデルマウス,および,心筋症を発症したのちにMYK-461を投与した肥大型心筋症モデルマウスとも,この200の遺伝子の発現はMYK-461を投与していない肥大型心筋症モデルマウスよりも野生型のマウスに類似していた.心筋症を発症したのちにMYK-461を投与した肥大型心筋症モデルマウスにおいては心筋の線維化の正常化は認められなかったものの,肥大型心筋症を発症したのちに心筋のサルコメアにおいて力の発生を抑制した場合,心筋細胞および線維芽細胞において遺伝子発現については正常化の傾向が誘発されたと考えられた.
心室壁の肥厚はエネルギー代謝の異常にも関与することから,ミトコンドリアタンパク質をコードする1158の遺伝子の発現について調べた.その結果,MYK-461を投与していない心筋症モデルマウスにおいてはミトコンドリアタンパク質をコードする遺伝子の20~30%に発現の異常があったが,MYK-461を投与した肥大型心筋症モデルマウスにおいては約5%の異常にとどまった.このことから,肥大型心筋症は代謝の異常と関連すること,そして,MYK-461の投与はこれを正常化することが示唆された.
おわりに
この研究において,筆者らは,心筋のサルコメアの仕事率を低下させる作用をもつ低分子阻害剤の早期からの投与が肥大型心筋症の形態学的および組織学的な異常,遺伝子発現の異常を改善することを示した.これは,肥大型心筋症の原因となる変異タンパク質が心筋のサルコメアの仕事率および収縮力を亢進し,結果として,病理的なリモデリングを促進するという説を支持すると考えられた.この結果は組換えタンパク質を用いた生体外におけるいくつかの実験の結果と相反するものであり,心筋のサルコメアを構成するあるいは仕事率の上昇に関与する重要なタンパク質が欠失している可能性のある生体外における実験系には限界のある可能性が示唆された.今回の研究は,ミオシンを直接の標的として力の発生を抑制する低分子阻害剤が生体において肥大型心筋症の病態を改善することを示し,心筋のサルコメアの仕事率を低下させることが肥大型心筋症の有力な治療法のひとつとなりうることが提唱された(図2).
拡張型心筋症あるいは心緻密化障害による心筋症など,ほかの遺伝性の心筋症においては,すでに心筋のサルコメアの仕事率の異常が報告されている8).今回,肥大型心筋症に対しても心筋のサルコメアの仕事率の正常化が病態の改善に貢献したという結果と考え合わせると,心筋のサルコメアの異常な仕事率を正常化することを目標とする薬剤の開発は心筋症の一般に共通した治療戦略として大いに期待できるものと考えられる.実際に,心不全の患者に対するミオシンの活性化薬の臨床治験はすでに開始されている.ヒトの遺伝子異常が心臓の機能にもたらす生化学的あるいは生理学的な効果につきさらに詳細に検討することにより,心筋症の根本的な発症の機序が解明され,将来的には根治療法につながることが期待される.
文 献
- Maron, B. J., Gardin, J. M., Flack, J. M. et al.: Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation, 92, 785-789 (1995)[PubMed]
- Ho, C. Y., Sweitzer, N. K., McDonough, B. et al.: Assessment of diastolic function with Doppler tissue imaging to predict genotype in preclinical hypertrophic cardiomyopathy. Circulation, 105, 2992-2997 (2002)[PubMed]
- Forsey, J., Benson, L., Rozenblyum, E. et al.: Early changes in apical rotation in genotype positive children with hypertrophic cardiomyopathy mutations without hypertrophic changes on two-dimensional imaging. J. Am. Soc. Echocardiogr., 27, 215-221 (2014)[PubMed]
- Russel, I. K., Brouwer, W. P., Germans, T. et al.: Increased left ventricular torsion in hypertrophic cardiomyopathy mutation carriers with normal wall thickness. J. Cardiovasc. Magn. Reson., 13, 3 (2011)[PubMed]
- Moore, J. R., Leinwand, L. & Warshaw, D. M.: Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circ. Res., 111, 375-385 (2012)[PubMed]
- Sommese, R. F., Sung, J., Nag, S. et al.: Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human β-cardiac myosin motor function. Proc. Natl. Acad. Sci. USA, 110, 12607-12612 (2013)[PubMed]
- Nag, S., Sommese, R. F., Ujfalusi, Z. et al.: Contractility parameters of human β-cardiac myosin with the hypertrophic cardiomyopathy mutation R403Q show loss of motor function. Sci. Adv., 1, e1500511 (2015)[PubMed]
- Aksel, T., Choe Yu, E., Sutton, S. et al.: Ensemble force changes that result from human cardiac myosin mutations and a small-molecule effector. Cell Rep., 11, 910-920 (2015)[PubMed]
著者プロフィール
略歴:2000年 東京医科歯科大学大学院医歯学総合研究科にて博士号取得,米国Boston Children’s Hospital研究員,2002年 東京医科歯科大学大学院医歯学総合研究科 助手,2006年 米国Boston Children’s Hospital研究員を経て,2007年より米国Harvard Medical School講師.
研究テーマ:遺伝性および発生の異常にもとづく心臓疾患の発生の機序.
関心事:働く女性のかかえる問題点の日米での比較検討および今後の展望.
© 2016 脇本 博子 Licensed under CC 表示 2.1 日本
