IDH1遺伝子の変異はニコチンアミドホスホリボシルトランスフェラーゼ阻害剤に対し著明な細胞脆弱性をもたらす
立石健祐・脇本浩明
(米国Harvard Medical School,Massachusetts General Hospital,Department of Neurosurgery)
email:立石健祐
DOI: 10.7875/first.author.2016.002
Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion.
Kensuke Tateishi, Hiroaki Wakimoto, A. John Iafrate, Shota Tanaka, Franziska Loebel, Nina Lelic, Dmitri Wiederschain, Olivier Bedel, Gejing Deng, Bailin Zhang, Timothy He, Xu Shi, Robert E. Gerszten, Yiyun Zhang, Jing-Ruey J. Yeh, William T. Curry, Dan Zhao, Sudhandra Sundaram, Fares Nigim, Mara V.A. Koerner, Quan Ho, David E. Fisher, Elisabeth M. Roider, Lajos V. Kemeny, Yardena Samuels, Keith T. Flaherty, Tracy T. Batchelor, Andrew S. Chi, Daniel P. Cahill
Cancer Cell, 28, 773-784 (2015)
イソクエン酸デヒドロゲナーゼ1をコードするIDH1遺伝子に変異をもつ腫瘍においては2-ヒドロキシグルタル酸が増加し腫瘍の形成に深く関与する可能性が示唆されており,近年,2-ヒドロキシグルタル酸を標的とした治療法の開発が提唱されている.今回,筆者らは,IDH1遺伝子変異をもつ患者に由来する悪性のグリオーマ幹細胞を用いて,IDH1阻害剤による2-ヒドロキシグルタル酸の抑制および腫瘍の制御の可能性について検討した.IDH1阻害剤は2-ヒドロキシグルタル酸の産生を強力に抑制したにもかかわらず,腫瘍の制御効果は認められなかった.一方で,IDH1遺伝子変異をもつ細胞においてIDH1阻害剤を投与したのちにおける代謝産物を網羅的に解析することにより,治療の標的としてNAD+が同定された.NAD+の合成に重要なサルベージ経路の律速酵素であるニコチンアミドホスホリボシルトランスフェラーゼの特異的な阻害剤は,IDH1遺伝子変異をもつ腫瘍においてNAD+の枯渇をひき起こすとともに,in vitroおよびin vivoにおいて顕著な抗腫瘍効果を発揮した.また,IDH1遺伝子変異は別のサルベージ経路の律速酵素であるニコチン酸ホスホリボシルトランスフェラーゼの発現を抑制し,これにともなうNAD+の濃度の低下が,IDH1遺伝子変異をもつ細胞におけるニコチンアミドホスホリボシルトランスフェラーゼ阻害剤に対する感受性に寄与することが明らかにされた.これらの結果から,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤によるNAD+の合成の阻害は,IDH1遺伝子変異をもつ腫瘍に対する新たな治療法につながる可能性が示唆された.
近年,イソクエン酸デヒドロゲナーゼをコードするIDH1遺伝子およびIDH2遺伝子の変異が,グリオーマ,急性骨髄性白血病,血管免疫芽球性T細胞リンパ腫,軟骨肉腫,胆管がんにおいて同定された1,2).グリオーマや軟骨肉腫においてはおもにIDH1遺伝子の変異がみられる一方,急性骨髄性白血病や血管免疫芽球性T細胞リンパ腫においてはIDH2遺伝子の変異が主である.グリオーマにおけるIDH1遺伝子の変異は,原発性グリオブラストーマではまれであるが,アストロサイトーマ,オリゴデンドログリオーマ,続発性グリオブラストーマでは50~80%と高頻度に認められる2).これらの変異により,イソクエン酸デヒドロゲナーゼのもつイソクエン酸から2-オキソグルタル酸への触媒活性が阻害される一方,NADPHに依存して2-オキソグルタル酸から2-ヒドロキシグルタル酸が産生される3,4).この2-ヒドロキシグルタル酸はがん代謝産物と呼称され,IDH1遺伝子あるいはIDH2遺伝子に変異をもつ腫瘍の形成に大きく関与すると考えられている.2-ヒドロキシグルタル酸は2-オキソグルタル酸と拮抗することにより2-オキソグルタル酸に依存性のジオキシゲナーゼの活性を阻害し,腫瘍化を促進するとされている4,5).また,IDH1遺伝子変異をもつ細胞に特徴的な所見として,ゲノムの全体にわたる広範なDNAメチル化の亢進がある6).実験モデルにおいては,IDH1遺伝子変異あるいはIDH2遺伝子変異の強制発現および2-ヒドロキシグルタル酸の投与によりこのDNAメチル化の亢進がひき起こされることが明らかにされている7).このため,2-ヒドロキシグルタル酸を抑制することによりIDH1遺伝子変異をもつ腫瘍を制御する新たな治療法の可能性が提唱されている8).これら2-ヒドロキシグルタル酸に関連する効果にくわえ,近年,IDH1遺伝子変異は解糖系,TCA回路,グルタミン代謝に影響を及ぼすことが明らかにされている9).
この研究においては,IDH1遺伝子変異をもつ腫瘍に対する有力な治療法を開発するために,患者に由来するIDH1遺伝子変異をもつ細胞をおもに用いて2-ヒドロキシグルタル酸の抑制による腫瘍の制御について評価するとともに,細胞における代謝産物の網羅的な解析をとおして腫瘍代謝の側面からIDH1遺伝子変異をもつ腫瘍の治療の標的を探求した.
従来から筆者らは,患者に由来するIDH1遺伝子変異をもつグリオブラストーマ細胞の樹立に取り組んでおり,これまで,複数の細胞株を確立してきた10).そこで,これらの細胞株を用いてIDH1阻害剤が細胞増殖の抑制に寄与するかどうか検討した.IDH1遺伝子変異をもつ細胞にIDH1阻害剤を投与したのち,2-ヒドロキシグルタル酸の変動について評価した.その結果,測定したすべてのIDH1遺伝子変異をもつ細胞において,2-ヒドロキシグルタル酸の濃度の上昇,および,IDH1阻害剤の投与による2-ヒドロキシグルタル酸の濃度の著明な低下が確認された.しかしながら,検討したすべてのIDH1遺伝子変異をもつ細胞において,IDH1阻害剤の投与による細胞増殖の抑制は認められず,逆に,多くの細胞において増殖の軽度の促進が認められた.
in vivoにおけるIDH1阻害剤による2-ヒドロキシグルタル酸の抑制効果について検討した.患者に由来するIDH1遺伝子変異をもつグリオブラストーマ細胞をSCIDマウスの脳へ移植し,IDH1阻害剤を投与したところ,IDH1阻害剤の腫瘍への集積および腫瘍細胞における2-ヒドロキシグルタル酸の濃度の低下が確認された.しかしながら,IDH1阻害剤を連続的に投与してもマウスの生存期間は延長しなかった.また,IDH1阻害剤の投与によりエピゲノムの変化に影響は検出されなかった.長期間にわたる2-ヒドロキシグルタル酸の抑制がIDH1遺伝子変異により生じたエピゲノムの変化にどのような影響を及ぼすか検討するため,患者に由来するIDH1遺伝子変異をもつグリオブラストーマ細胞に対し1年にわたりIDH1阻害剤を継続的に投与した.その結果,2-ヒドロキシグルタル酸の長期にわたる抑制にもかかわらず,エピゲノムの変化に明らかな影響は認められなかった.さらに,有意な細胞増殖の促進がin vitroおよびin vivoにおいて確認された.
これらの結果から,2-ヒドロキシグルタル酸の抑制はIDH1遺伝子変異をもつ多くの腫瘍において細胞増殖の抑制には寄与しないこと,さらには,IDH1遺伝子変異により生じたエピゲノムの変化は容易には影響をうけないことが示唆された.
IDH1遺伝子変異をもつ腫瘍における代謝の特異性を明らかにし治療の新たな標的を同定するため,患者に由来するIDH1遺伝子変異をもつグリオブラストーマ細胞にIDH1阻害剤を投与したのち,細胞における代謝産物の変化を液体クロマトグラフィー-質量分析法により網羅的に解析した.短期間および長期間の投与により2-ヒドロキシグルタル酸の著明な抑制が認められた一方,50%以上の上昇が認められた代謝産物として,NADH,クエン酸,グリセロール3-リン酸が検出された.また,別の解析手法により,NADHのみならずNAD+もIDH1阻害剤を投与したのち上昇することが判明した.NAD+を標的とした治療は一部の腫瘍モデルにおいて有効性が示されていたことから,NAD+について,
IDH1遺伝子変異をもつ腫瘍に対する治療の新たな標的としての可能性を探索することにした.
患者に由来するIDH1遺伝子変異をもつグリオブラストーマ細胞を用いて,NAD+の合成において主要なサルベージ経路の律速酵素であるニコチンアミドホスホリボシルトランスフェラーゼの阻害剤に対する感受性について評価した.その結果,解析したすべてのIDH1遺伝子変異をもつ細胞に対しては数nMレベルと非常に強力な細胞毒性をもつ一方,野生型の細胞は耐性を示した.ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤の投与によるNAD+の変化について評価したところ,IDH1遺伝子変異をもつ細胞においてはNAD+の濃度は著明に低下していた.薬理作用について検討するため,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤と同時に下流の代謝産物であるニコチンアミドモノヌクレオチドを投与したところ,細胞毒性はまったく示されずNAD+の濃度も回復した.さらに,NAD+の投与によってもニコチンアミドホスホリボシルトランスフェラーゼ阻害薬による細胞毒性は示されなくなったことから,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤はニコチンアミドホスホリボシルトランスフェラーゼを直接に抑制することによりNAD+の合成を阻害し,NAD+が致死性の閾値以下に枯渇することにより細胞毒性の生じることが判明した.
なぜ,ニコチンアミドホスホリボシルトランスフェラーゼの阻害剤がIDH1遺伝子変異をもつ細胞に対し細胞毒性を示すのか検討した.細胞におけるNAD+の基礎的な濃度を患者に由来するIDH1遺伝子変異をもつ細胞と野生型の細胞とで比較したところ,IDH1遺伝子変異をもつ細胞において有意にNAD+の濃度が低いことが判明した.NAD+の合成に重要なサルベージ経路としては,ニコチンアミドホスホリボシルトランスフェラーゼを律速酵素とする経路のほか,ニコチン酸ホスホリボシルトランスフェラーゼを律速酵素とする経路がある.ニコチンアミドホスホリボシルトランスフェラーゼとニコチン酸ホスホリボシルトランスフェラーゼの発現を比較したところ,ニコチンアミドホスホリボシルトランスフェラーゼはすべての腫瘍細胞において発現していたのに対し,IDH1遺伝子変異をもつ細胞においてはニコチン酸ホスホリボシルトランスフェラーゼの発現は著明に低下していた.また,ニコチン酸ホスホリボシルトランスフェラーゼの上流の代謝産物であるニコチン酸を投与しても,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤による細胞毒性は回復しなかった.
IDH1遺伝子変異がニコチン酸ホスホリボシルトランスフェラーゼの発現に及ぼす影響について評価するため,野生型のグリオブラストーマ細胞を用いてIDH1遺伝子変異の強制発現モデルを作製した.その結果,IDH1遺伝子変異の強制発現によりニコチン酸ホスホリボシルトランスフェラーゼの発現は著明に低下し,NAD+の濃度の低下とともにニコチンアミドホスホリボシルトランスフェラーゼ阻害剤に対する感受性が上昇した.さらに,IDH1遺伝子変異をもつグリオブラストーマ細胞においてニコチン酸ホスホリボシルトランスフェラーゼを強制発現したところ,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤に対する強い耐性が認められた.また,ニコチン酸ホスホリボシルトランスフェラーゼをコードするNAPRT1遺伝子のプロモーター領域のDNAメチル化について解析したところ,ニコチン酸ホスホリボシルトランスフェラーゼの発現が低い細胞において高いDNAメチル化が認められた.これらの結果から,IDH1遺伝子変異はNAPRT1遺伝子のプロモーター領域のDNAメチル化を介してニコチン酸ホスホリボシルトランスフェラーゼの発現を抑制し,その結果,NAD+の濃度は低下することが示唆された.そのため,IDH1遺伝子変異をもつ細胞においてはニコチンアミドホスホリボシルトランスフェラーゼ阻害剤の投与によりNAD+が枯渇しすみやかに致死性の閾値に達することにより細胞死が生じるものと考えられた.
IDH1遺伝子変異はTCA回路における酸化的リン酸化を促進させることが知られている9).そこで,患者に由来するIDH1遺伝子変異をもつグリオブラストーマ細胞を用いて,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤がTCA回路に及ぼす影響について検討した.その結果,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤によるNAD+の枯渇は,IDH1遺伝子変異に対しより高い依存度を示すTCA回路の機能を破綻させることが判明した.
ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤はIDH1遺伝子変異をもつ細胞においてATPの濃度を著明に低下させ,そののち,すみやかに細胞死の生じることが判明した.ATPの濃度の低下はNAD+の枯渇による酸化的リン酸化の破綻によるものと推察された.
細胞死の機構について明らかにするため,アポトーシス経路について評価したが有意な活性化は認められなかった.一方,細胞死の機構としてオートファジーの関与がうたがわれた.一般に,ATPの濃度の低下によりAMPキナーゼが活性化することは広く知られている11).患者に由来するIDH1遺伝子変異をもつグリオブラストーマ細胞において,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤はAMPキナーゼのリン酸化を介して,オートファジーを促進するULK1のSer555およびAtg13のリン酸化を促進し,mTORのリン酸化およびその下流の4E-BP1,S6のリン酸化を抑制するとともに,リン酸化によりオートファジーを抑制するULK1のSer757の脱リン酸化を促進した.これらのシグナルの変化はニコチンアミドホスホリボシルトランスフェラーゼの下流の代謝産物であるニコチンアミドモノヌクレオチドの投与により部分的に拮抗されたことからも,オートファジーはニコチンアミドホスホリボシルトランスフェラーゼ阻害剤の投与による直接的な影響と考えられた.また,NAD+の枯渇によるAMPキナーゼ経路の活性化が細胞死の主要な機構であることが示された.
in vivoにおいて患者に由来するIDH1遺伝子変異をもつ腫瘍に対するニコチンアミドホスホリボシルトランスフェラーゼ阻害剤の効果を検討した.SCIDマウスの皮下にIDH1遺伝子変異をもつフィブロサルコーマ細胞を移植したのち,連日,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤を投与したところ,対照となるマウスと比較して腫瘍の増大が顕著に抑制された.また,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤を投与したマウスの腫瘍組織においては細胞増殖マーカーの発現が著明に低下していた.
脳腫瘍における評価のため,SCIDマウスの脳に患者に由来するIDH1遺伝子変異をもつグリオブラストーマ細胞を移植したのち,週1回,高容量のニコチンアミドホスホリボシルトランスフェラーゼ阻害剤を全身投与した.その結果,生存期間の有意な延長が認められた.また,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤を投与したのち,脳の腫瘍組織においてNAD+を測定したところほぼ枯渇しており,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤の脳腫瘍への移行が確認された.体重の減少など明らかな有害事象は認められなかった.
これらの結果から,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤はin vivoにおいてもIDH1遺伝子変異をもつ腫瘍に対する有効な治療法であることが判明した.
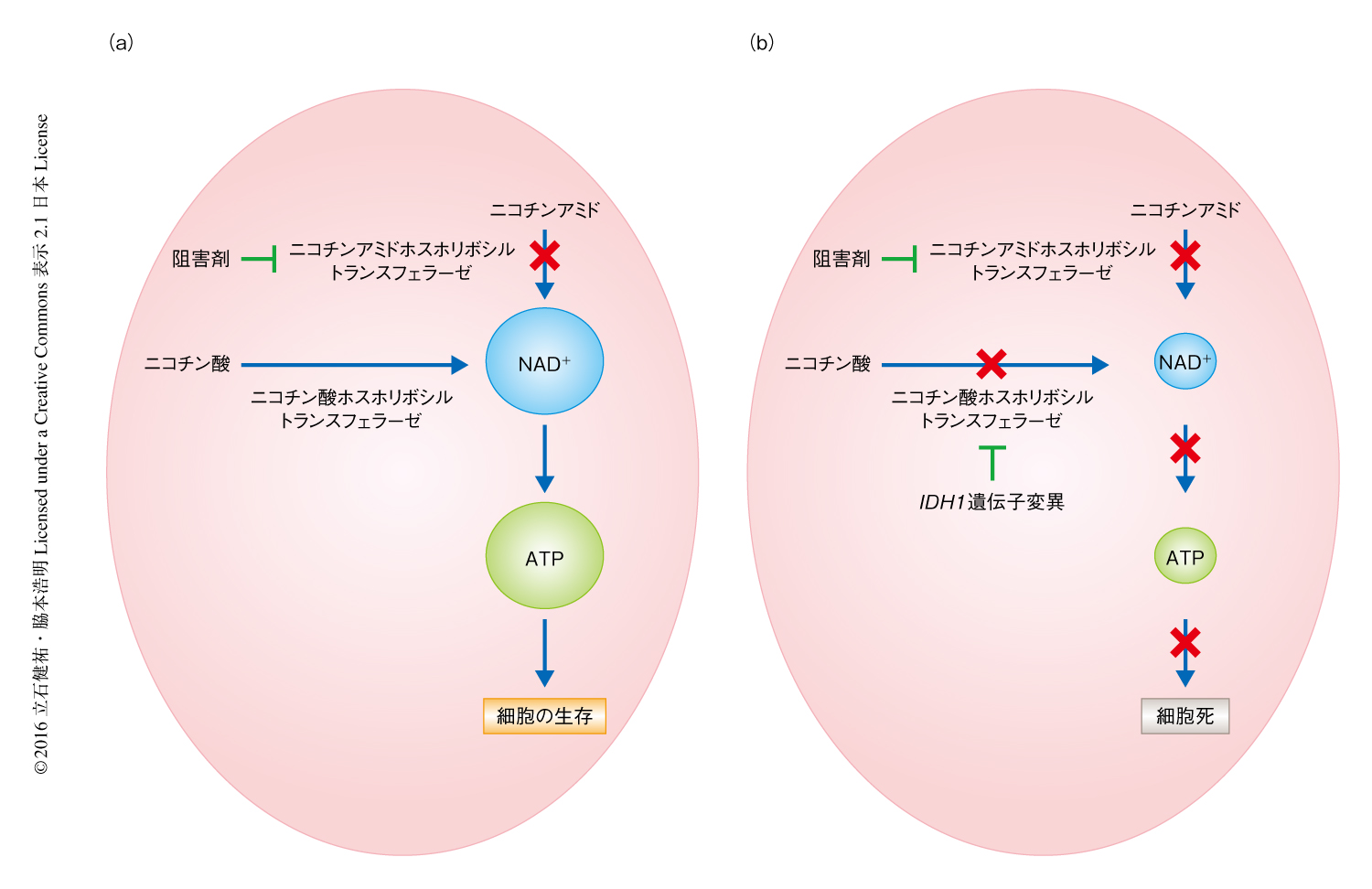
患者に由来するIDH1遺伝子変異をもつグリオブラストーマを中心に検討した今回の研究により,2-ヒドロキシグルタル酸の抑制はIDH1遺伝子変異をもつ腫瘍の制御には必ずしも寄与しないことが示された.また,IDH1遺伝子変異により生じたエピゲノム変化は長期間にわたる2-ヒドロキシグルタル酸の抑制によっても影響されないことが判明した.近年,2-ヒドロキシグルタル酸の抑制はIDH1遺伝子変異をもつ腫瘍の治療の標的として有力視されていたが,今回の研究は,たんに2-ヒドロキシグルタル酸を抑制するだけでは治療効果の得られないIDH1遺伝子変異をもつ腫瘍が存在することを明らかにした点で意義深い.一方で,IDH1遺伝子変異によるNAD+の代謝の変化が明らかにされた.IDH1遺伝子変異はNAD+の合成に重要なサルベージ経路の律速酵素であるニコチン酸ホスホリボシルトランスフェラーゼを抑制することによりNAD+の濃度を低下させる.これにより,別のサルベージ経路の律速酵素であるニコチンアミドホスホリボシルトランスフェラーゼを標的とした治療に対し,著明な細胞脆弱性を示すことが明らかにされた(図1).これらの結果は,今後,NAD+を標的とした治療がIDH1遺伝子変異をもつ腫瘍に対する有力な治療法になりうる可能性を示した.欧米においては,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤のがん治療における臨床治験が進行しており,悪性の脳腫瘍を含めたIDH1遺伝子変異をもつ腫瘍に対する臨床応用が治療成績の向上につながることが期待される.
略歴:2013年 横浜市立大学大学院医学研究科にて博士号取得,同年より米国Harvard Medical Schoolポスドク.
抱負:脳腫瘍の患者への還元を合言葉に,臨床医と基礎研究者の両輪で活動することを目標にしている.また,臨床医の強みを活かした研究テーマを模索している.
脇本 浩明(Hiroaki Wakimoto)
米国Harvard Medical SchoolにてAssistant Professor.
© 2016 立石健祐・脇本浩明 Licensed under CC 表示 2.1 日本
(米国Harvard Medical School,Massachusetts General Hospital,Department of Neurosurgery)
email:立石健祐
DOI: 10.7875/first.author.2016.002
Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion.
Kensuke Tateishi, Hiroaki Wakimoto, A. John Iafrate, Shota Tanaka, Franziska Loebel, Nina Lelic, Dmitri Wiederschain, Olivier Bedel, Gejing Deng, Bailin Zhang, Timothy He, Xu Shi, Robert E. Gerszten, Yiyun Zhang, Jing-Ruey J. Yeh, William T. Curry, Dan Zhao, Sudhandra Sundaram, Fares Nigim, Mara V.A. Koerner, Quan Ho, David E. Fisher, Elisabeth M. Roider, Lajos V. Kemeny, Yardena Samuels, Keith T. Flaherty, Tracy T. Batchelor, Andrew S. Chi, Daniel P. Cahill
Cancer Cell, 28, 773-784 (2015)
要 約
イソクエン酸デヒドロゲナーゼ1をコードするIDH1遺伝子に変異をもつ腫瘍においては2-ヒドロキシグルタル酸が増加し腫瘍の形成に深く関与する可能性が示唆されており,近年,2-ヒドロキシグルタル酸を標的とした治療法の開発が提唱されている.今回,筆者らは,IDH1遺伝子変異をもつ患者に由来する悪性のグリオーマ幹細胞を用いて,IDH1阻害剤による2-ヒドロキシグルタル酸の抑制および腫瘍の制御の可能性について検討した.IDH1阻害剤は2-ヒドロキシグルタル酸の産生を強力に抑制したにもかかわらず,腫瘍の制御効果は認められなかった.一方で,IDH1遺伝子変異をもつ細胞においてIDH1阻害剤を投与したのちにおける代謝産物を網羅的に解析することにより,治療の標的としてNAD+が同定された.NAD+の合成に重要なサルベージ経路の律速酵素であるニコチンアミドホスホリボシルトランスフェラーゼの特異的な阻害剤は,IDH1遺伝子変異をもつ腫瘍においてNAD+の枯渇をひき起こすとともに,in vitroおよびin vivoにおいて顕著な抗腫瘍効果を発揮した.また,IDH1遺伝子変異は別のサルベージ経路の律速酵素であるニコチン酸ホスホリボシルトランスフェラーゼの発現を抑制し,これにともなうNAD+の濃度の低下が,IDH1遺伝子変異をもつ細胞におけるニコチンアミドホスホリボシルトランスフェラーゼ阻害剤に対する感受性に寄与することが明らかにされた.これらの結果から,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤によるNAD+の合成の阻害は,IDH1遺伝子変異をもつ腫瘍に対する新たな治療法につながる可能性が示唆された.
はじめに
近年,イソクエン酸デヒドロゲナーゼをコードするIDH1遺伝子およびIDH2遺伝子の変異が,グリオーマ,急性骨髄性白血病,血管免疫芽球性T細胞リンパ腫,軟骨肉腫,胆管がんにおいて同定された1,2).グリオーマや軟骨肉腫においてはおもにIDH1遺伝子の変異がみられる一方,急性骨髄性白血病や血管免疫芽球性T細胞リンパ腫においてはIDH2遺伝子の変異が主である.グリオーマにおけるIDH1遺伝子の変異は,原発性グリオブラストーマではまれであるが,アストロサイトーマ,オリゴデンドログリオーマ,続発性グリオブラストーマでは50~80%と高頻度に認められる2).これらの変異により,イソクエン酸デヒドロゲナーゼのもつイソクエン酸から2-オキソグルタル酸への触媒活性が阻害される一方,NADPHに依存して2-オキソグルタル酸から2-ヒドロキシグルタル酸が産生される3,4).この2-ヒドロキシグルタル酸はがん代謝産物と呼称され,IDH1遺伝子あるいはIDH2遺伝子に変異をもつ腫瘍の形成に大きく関与すると考えられている.2-ヒドロキシグルタル酸は2-オキソグルタル酸と拮抗することにより2-オキソグルタル酸に依存性のジオキシゲナーゼの活性を阻害し,腫瘍化を促進するとされている4,5).また,IDH1遺伝子変異をもつ細胞に特徴的な所見として,ゲノムの全体にわたる広範なDNAメチル化の亢進がある6).実験モデルにおいては,IDH1遺伝子変異あるいはIDH2遺伝子変異の強制発現および2-ヒドロキシグルタル酸の投与によりこのDNAメチル化の亢進がひき起こされることが明らかにされている7).このため,2-ヒドロキシグルタル酸を抑制することによりIDH1遺伝子変異をもつ腫瘍を制御する新たな治療法の可能性が提唱されている8).これら2-ヒドロキシグルタル酸に関連する効果にくわえ,近年,IDH1遺伝子変異は解糖系,TCA回路,グルタミン代謝に影響を及ぼすことが明らかにされている9).
この研究においては,IDH1遺伝子変異をもつ腫瘍に対する有力な治療法を開発するために,患者に由来するIDH1遺伝子変異をもつ細胞をおもに用いて2-ヒドロキシグルタル酸の抑制による腫瘍の制御について評価するとともに,細胞における代謝産物の網羅的な解析をとおして腫瘍代謝の側面からIDH1遺伝子変異をもつ腫瘍の治療の標的を探求した.
1.IDH1阻害剤はIDH1遺伝子変異をもつ細胞において2-ヒドロキシグルタル酸を抑制するが細胞増殖は抑制しない
従来から筆者らは,患者に由来するIDH1遺伝子変異をもつグリオブラストーマ細胞の樹立に取り組んでおり,これまで,複数の細胞株を確立してきた10).そこで,これらの細胞株を用いてIDH1阻害剤が細胞増殖の抑制に寄与するかどうか検討した.IDH1遺伝子変異をもつ細胞にIDH1阻害剤を投与したのち,2-ヒドロキシグルタル酸の変動について評価した.その結果,測定したすべてのIDH1遺伝子変異をもつ細胞において,2-ヒドロキシグルタル酸の濃度の上昇,および,IDH1阻害剤の投与による2-ヒドロキシグルタル酸の濃度の著明な低下が確認された.しかしながら,検討したすべてのIDH1遺伝子変異をもつ細胞において,IDH1阻害剤の投与による細胞増殖の抑制は認められず,逆に,多くの細胞において増殖の軽度の促進が認められた.
in vivoにおけるIDH1阻害剤による2-ヒドロキシグルタル酸の抑制効果について検討した.患者に由来するIDH1遺伝子変異をもつグリオブラストーマ細胞をSCIDマウスの脳へ移植し,IDH1阻害剤を投与したところ,IDH1阻害剤の腫瘍への集積および腫瘍細胞における2-ヒドロキシグルタル酸の濃度の低下が確認された.しかしながら,IDH1阻害剤を連続的に投与してもマウスの生存期間は延長しなかった.また,IDH1阻害剤の投与によりエピゲノムの変化に影響は検出されなかった.長期間にわたる2-ヒドロキシグルタル酸の抑制がIDH1遺伝子変異により生じたエピゲノムの変化にどのような影響を及ぼすか検討するため,患者に由来するIDH1遺伝子変異をもつグリオブラストーマ細胞に対し1年にわたりIDH1阻害剤を継続的に投与した.その結果,2-ヒドロキシグルタル酸の長期にわたる抑制にもかかわらず,エピゲノムの変化に明らかな影響は認められなかった.さらに,有意な細胞増殖の促進がin vitroおよびin vivoにおいて確認された.
これらの結果から,2-ヒドロキシグルタル酸の抑制はIDH1遺伝子変異をもつ多くの腫瘍において細胞増殖の抑制には寄与しないこと,さらには,IDH1遺伝子変異により生じたエピゲノムの変化は容易には影響をうけないことが示唆された.
2.IDH1遺伝子変異をもつ腫瘍の治療の標的としてNAD+が同定された
IDH1遺伝子変異をもつ腫瘍における代謝の特異性を明らかにし治療の新たな標的を同定するため,患者に由来するIDH1遺伝子変異をもつグリオブラストーマ細胞にIDH1阻害剤を投与したのち,細胞における代謝産物の変化を液体クロマトグラフィー-質量分析法により網羅的に解析した.短期間および長期間の投与により2-ヒドロキシグルタル酸の著明な抑制が認められた一方,50%以上の上昇が認められた代謝産物として,NADH,クエン酸,グリセロール3-リン酸が検出された.また,別の解析手法により,NADHのみならずNAD+もIDH1阻害剤を投与したのち上昇することが判明した.NAD+を標的とした治療は一部の腫瘍モデルにおいて有効性が示されていたことから,NAD+について,
IDH1遺伝子変異をもつ腫瘍に対する治療の新たな標的としての可能性を探索することにした.
3.ニコチンアミドホスホリボシルトランスフェラーゼの阻害剤はIDH1遺伝子変異をもつ腫瘍に対し強力な細胞毒性を示す
患者に由来するIDH1遺伝子変異をもつグリオブラストーマ細胞を用いて,NAD+の合成において主要なサルベージ経路の律速酵素であるニコチンアミドホスホリボシルトランスフェラーゼの阻害剤に対する感受性について評価した.その結果,解析したすべてのIDH1遺伝子変異をもつ細胞に対しては数nMレベルと非常に強力な細胞毒性をもつ一方,野生型の細胞は耐性を示した.ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤の投与によるNAD+の変化について評価したところ,IDH1遺伝子変異をもつ細胞においてはNAD+の濃度は著明に低下していた.薬理作用について検討するため,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤と同時に下流の代謝産物であるニコチンアミドモノヌクレオチドを投与したところ,細胞毒性はまったく示されずNAD+の濃度も回復した.さらに,NAD+の投与によってもニコチンアミドホスホリボシルトランスフェラーゼ阻害薬による細胞毒性は示されなくなったことから,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤はニコチンアミドホスホリボシルトランスフェラーゼを直接に抑制することによりNAD+の合成を阻害し,NAD+が致死性の閾値以下に枯渇することにより細胞毒性の生じることが判明した.
4.IDH1遺伝子変異をもつ細胞はニコチン酸ホスホリボシルトランスフェラーゼが抑制されることによりNAD+の濃度が低下する
なぜ,ニコチンアミドホスホリボシルトランスフェラーゼの阻害剤がIDH1遺伝子変異をもつ細胞に対し細胞毒性を示すのか検討した.細胞におけるNAD+の基礎的な濃度を患者に由来するIDH1遺伝子変異をもつ細胞と野生型の細胞とで比較したところ,IDH1遺伝子変異をもつ細胞において有意にNAD+の濃度が低いことが判明した.NAD+の合成に重要なサルベージ経路としては,ニコチンアミドホスホリボシルトランスフェラーゼを律速酵素とする経路のほか,ニコチン酸ホスホリボシルトランスフェラーゼを律速酵素とする経路がある.ニコチンアミドホスホリボシルトランスフェラーゼとニコチン酸ホスホリボシルトランスフェラーゼの発現を比較したところ,ニコチンアミドホスホリボシルトランスフェラーゼはすべての腫瘍細胞において発現していたのに対し,IDH1遺伝子変異をもつ細胞においてはニコチン酸ホスホリボシルトランスフェラーゼの発現は著明に低下していた.また,ニコチン酸ホスホリボシルトランスフェラーゼの上流の代謝産物であるニコチン酸を投与しても,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤による細胞毒性は回復しなかった.
IDH1遺伝子変異がニコチン酸ホスホリボシルトランスフェラーゼの発現に及ぼす影響について評価するため,野生型のグリオブラストーマ細胞を用いてIDH1遺伝子変異の強制発現モデルを作製した.その結果,IDH1遺伝子変異の強制発現によりニコチン酸ホスホリボシルトランスフェラーゼの発現は著明に低下し,NAD+の濃度の低下とともにニコチンアミドホスホリボシルトランスフェラーゼ阻害剤に対する感受性が上昇した.さらに,IDH1遺伝子変異をもつグリオブラストーマ細胞においてニコチン酸ホスホリボシルトランスフェラーゼを強制発現したところ,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤に対する強い耐性が認められた.また,ニコチン酸ホスホリボシルトランスフェラーゼをコードするNAPRT1遺伝子のプロモーター領域のDNAメチル化について解析したところ,ニコチン酸ホスホリボシルトランスフェラーゼの発現が低い細胞において高いDNAメチル化が認められた.これらの結果から,IDH1遺伝子変異はNAPRT1遺伝子のプロモーター領域のDNAメチル化を介してニコチン酸ホスホリボシルトランスフェラーゼの発現を抑制し,その結果,NAD+の濃度は低下することが示唆された.そのため,IDH1遺伝子変異をもつ細胞においてはニコチンアミドホスホリボシルトランスフェラーゼ阻害剤の投与によりNAD+が枯渇しすみやかに致死性の閾値に達することにより細胞死が生じるものと考えられた.
5.IDH1遺伝子変異をもつ細胞においてニコチンアミドホスホリボシルトランスフェラーゼ阻害剤はNAD+の枯渇によりTCA回路を破綻させる
IDH1遺伝子変異はTCA回路における酸化的リン酸化を促進させることが知られている9).そこで,患者に由来するIDH1遺伝子変異をもつグリオブラストーマ細胞を用いて,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤がTCA回路に及ぼす影響について検討した.その結果,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤によるNAD+の枯渇は,IDH1遺伝子変異に対しより高い依存度を示すTCA回路の機能を破綻させることが判明した.
6.IDH1遺伝子変異をもつ細胞はNAD+の枯渇によりオートファジーによる細胞死をひき起こす
ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤はIDH1遺伝子変異をもつ細胞においてATPの濃度を著明に低下させ,そののち,すみやかに細胞死の生じることが判明した.ATPの濃度の低下はNAD+の枯渇による酸化的リン酸化の破綻によるものと推察された.
細胞死の機構について明らかにするため,アポトーシス経路について評価したが有意な活性化は認められなかった.一方,細胞死の機構としてオートファジーの関与がうたがわれた.一般に,ATPの濃度の低下によりAMPキナーゼが活性化することは広く知られている11).患者に由来するIDH1遺伝子変異をもつグリオブラストーマ細胞において,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤はAMPキナーゼのリン酸化を介して,オートファジーを促進するULK1のSer555およびAtg13のリン酸化を促進し,mTORのリン酸化およびその下流の4E-BP1,S6のリン酸化を抑制するとともに,リン酸化によりオートファジーを抑制するULK1のSer757の脱リン酸化を促進した.これらのシグナルの変化はニコチンアミドホスホリボシルトランスフェラーゼの下流の代謝産物であるニコチンアミドモノヌクレオチドの投与により部分的に拮抗されたことからも,オートファジーはニコチンアミドホスホリボシルトランスフェラーゼ阻害剤の投与による直接的な影響と考えられた.また,NAD+の枯渇によるAMPキナーゼ経路の活性化が細胞死の主要な機構であることが示された.
7.ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤はマウスにおいてIDH1遺伝子変異をもつ腫瘍を強力に抑制する
in vivoにおいて患者に由来するIDH1遺伝子変異をもつ腫瘍に対するニコチンアミドホスホリボシルトランスフェラーゼ阻害剤の効果を検討した.SCIDマウスの皮下にIDH1遺伝子変異をもつフィブロサルコーマ細胞を移植したのち,連日,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤を投与したところ,対照となるマウスと比較して腫瘍の増大が顕著に抑制された.また,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤を投与したマウスの腫瘍組織においては細胞増殖マーカーの発現が著明に低下していた.
脳腫瘍における評価のため,SCIDマウスの脳に患者に由来するIDH1遺伝子変異をもつグリオブラストーマ細胞を移植したのち,週1回,高容量のニコチンアミドホスホリボシルトランスフェラーゼ阻害剤を全身投与した.その結果,生存期間の有意な延長が認められた.また,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤を投与したのち,脳の腫瘍組織においてNAD+を測定したところほぼ枯渇しており,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤の脳腫瘍への移行が確認された.体重の減少など明らかな有害事象は認められなかった.
これらの結果から,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤はin vivoにおいてもIDH1遺伝子変異をもつ腫瘍に対する有効な治療法であることが判明した.
おわりに
患者に由来するIDH1遺伝子変異をもつグリオブラストーマを中心に検討した今回の研究により,2-ヒドロキシグルタル酸の抑制はIDH1遺伝子変異をもつ腫瘍の制御には必ずしも寄与しないことが示された.また,IDH1遺伝子変異により生じたエピゲノム変化は長期間にわたる2-ヒドロキシグルタル酸の抑制によっても影響されないことが判明した.近年,2-ヒドロキシグルタル酸の抑制はIDH1遺伝子変異をもつ腫瘍の治療の標的として有力視されていたが,今回の研究は,たんに2-ヒドロキシグルタル酸を抑制するだけでは治療効果の得られないIDH1遺伝子変異をもつ腫瘍が存在することを明らかにした点で意義深い.一方で,IDH1遺伝子変異によるNAD+の代謝の変化が明らかにされた.IDH1遺伝子変異はNAD+の合成に重要なサルベージ経路の律速酵素であるニコチン酸ホスホリボシルトランスフェラーゼを抑制することによりNAD+の濃度を低下させる.これにより,別のサルベージ経路の律速酵素であるニコチンアミドホスホリボシルトランスフェラーゼを標的とした治療に対し,著明な細胞脆弱性を示すことが明らかにされた(図1).これらの結果は,今後,NAD+を標的とした治療がIDH1遺伝子変異をもつ腫瘍に対する有力な治療法になりうる可能性を示した.欧米においては,ニコチンアミドホスホリボシルトランスフェラーゼ阻害剤のがん治療における臨床治験が進行しており,悪性の脳腫瘍を含めたIDH1遺伝子変異をもつ腫瘍に対する臨床応用が治療成績の向上につながることが期待される.
文 献
- Amary, M. F., Bacsi, K., Maggiani, F. et al.: IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol., 224, 334-343 (2011)[PubMed]
- Yan, H., Parsons, D. W., Jin, G. et al.: IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med., 360, 765-773 (2009)[PubMed]
- Dang, L., White, D. W., Gross, S. et al.: Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature, 462, 739-744 (2009)[PubMed]
- Zhao, S., Lin, Y., Xu, W. et al.: Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science, 324, 261-265 (2009)[PubMed]
- Krall, A. S. & Christofk, H. R.: Cancer: a metabolic metamorphosis. Nature, 496, 38-40 (2013)[PubMed]
- Noushmehr, H., Weisenberger, D. J., Diefes, K. et al.: Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell, 17, 510-522 (2010)[PubMed]
- Turcan, S., Rohle, D., Goenka, A. et al.: IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature, 483, 479-483 (2012)[PubMed]
- Rohle, D., Popovici-Muller, J., Palaskas, N. et al.: An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science, 340, 626-630 (2013)[PubMed]
- Grassian, A. R., Parker, S. J., Davidson, S. M. et al.: IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res., 74, 3317-3331 (2014)[PubMed]
- Wakimoto, H., Tanaka, S., Curry, W. T. et al.: Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clin. Cancer Res., 20, 2898-2909 (2014)[PubMed]
- Hardie, D. G.: AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol., 8, 774-785 (2007)[PubMed]
著者プロフィール
略歴:2013年 横浜市立大学大学院医学研究科にて博士号取得,同年より米国Harvard Medical Schoolポスドク.
抱負:脳腫瘍の患者への還元を合言葉に,臨床医と基礎研究者の両輪で活動することを目標にしている.また,臨床医の強みを活かした研究テーマを模索している.
脇本 浩明(Hiroaki Wakimoto)
米国Harvard Medical SchoolにてAssistant Professor.
© 2016 立石健祐・脇本浩明 Licensed under CC 表示 2.1 日本
