SCFβ-TRCPユビキチンリガーゼ複合体によるリン酸化依存的なMdm2の分解制御機構の解明
犬塚博之・Wenyi Wei
(米国Harvard大学Medical School,Beth Israel Deaconess Medical Center,Department of Pathology)
email:犬塚博之
DOI: 10.7875/first.author.2010.018
Phosphorylation by casein kinase I promotes the turnover of the Mdm2 oncoprotein via the SCFβ-TRCP ubiquitin ligase.
Hiroyuki Inuzuka, Alan Tseng, Daming Gao, Bo Zhai, Qing Zhang, Shavali Shaik, Lixin Wan, Xiaolu L. Ang, Caroline Mock, Haoqiang Yin, Jayne M. Stommel, Steven Gygi, Galit Lahav, John Asara, Zhi-Xiong Jim Xiao, William G. Kaelin, J. Wade Harper, Wenyi Wei
Cancer Cell, 18, 147-159 (2010)
Mdm2はユビキチンリガーゼとしてがん抑制遺伝子産物p53の量的な制御をつかさどる重要なタンパク質であり,p53の活性はMdm2の発現量に応じた負の制御をうけている.Mdm2自体もユビキチン-プロテアソーム系による分解を介する発現制御をうけることが示されているが,その分子機構に関する知見は少ない.今回,筆者らは,DNA損傷により誘導されるすみやかなMdm2の分解が,カゼインキナーゼIによるMdm2のリン酸化と,それにつづくSCFβ-TRCPユビキチンリガーゼのMdm2への結合,そして,ユビキチン化,という過程により制御されていることを見い出した.さらに,SCFβ-TRCPによるMdm2の分解制御が,DNA損傷の際に出現するp53発現量の振動を直接的に制御していることも明らかにした.今回の報告は,カゼインキナーゼI-SCFβ-TRCP経路を介するMdm2の分解経路の破綻が発がんリスクを高める可能性を示唆するものであり,がんの分野の中心課題であるp53シグナル解析に新たな知見をもたらすものと期待される.
多くのヒト腫瘍においては,がん遺伝子の活性化に付随して起こる主要ながん抑制遺伝子の機能欠損が細胞のがん化をひき起こしている.p53はもっとも重要ながん抑制遺伝子産物のひとつであり,ヒト腫瘍の半数近くにおいて変異が報告されている1).細胞がDNA損傷などのストレスを感知すると,p53はすみやかに,細胞周期の停止やアポトーシス,DNA修復などに関連する遺伝子の発現を誘導して,細胞のがん化を防いでいる.その一方で,p53の異常な活性化は予期せぬアポトーシスや細胞老化をまねくことから,p53の活性制御には非常に緻密で幾重もの制御機構が存在する.
p53のおもな制御機構としては,翻訳後修飾のほか,その量的な制御がある.ストレス非存在下の細胞においてp53の発現量は基底レベルに抑えられているが,その制御はおもにユビキチンリガーゼであるMdm2が担っている2).Mdm2はp53に結合してそのユビキチン化を行い,プロテアソームによるp53の分解を促進している.そして,いったん細胞がDNA損傷シグナルを感知すると,Mdm2はすみやかに発現量を減少・消失させることによってp53を安定化させ,p53の活性を上昇させる.したがって,Mdm2の分解制御機構はp53活性化を直接的に制御するうえで非常に重要であるが,Mdm2分解の分子機序については未解明な点が多い.当初の知見では,自己ユビキチン化がその主要な分子機構であると考えられていたが3),近年,自己ユビキチン化能を消失させたMdm2変異体を発現するノックインマウスにおいても,依然として,このMdm2変異体がユビキチン-プロテアソーム系を介して分解をうけていることが明らかとなった4).このことは,Mdm2が自己ユビキチン化以外に,未同定のユビキチンリガーゼによってユビキチン化をうけ,その発現量を変動させていることを示唆している.
このような背景のもと,この研究では,Mdm2の分解制御機構の分子基盤の解明のため,Mdm2のユビキチン化を促進するタンパク質の同定と,それらのp53活性化への関与を分子レベルで明らかにすることを主要な目的とした.
現在,報告もしくは存在が推定されているユビキチンリガーゼのなかでCullin-RINGフィンガー型ユビキチンリガーゼが最大のファミリーを形成しており,その数は数百にも及ぶと推定されている5).このファミリーに属するユビキチンリガーゼがMdm2の分解をつかさどっている可能性を考え,最初に,免疫沈降法により細胞内でのCullinとMdm2との相互作用の検出を試みた.そして,個々のCullinファミリータンパク質の強制発現において,Mdm2がそのファミリーのひとつCullin1とのみ特異的に相互作用することを見い出した.Cullin1はユビキチンリガーゼ複合体のなかで足場タンパク質として機能し,現在,70種類以上も報告されているFボックスタンパク質とよばれる可変的な基質認識サブユニットと,アダプタータンパク質Skp1を介して選択的に結合することで,基質認識の多様性を生み出している.これら,Skp1,Cullin1,Fボックスタンパク質からなるユビキチンリガーゼ複合体はSCF(Skp1-Cullin1-F-box protein)複合体とよばれている6).
細胞周期におけるMdm2の発現解析から,Mdm2はG1/S遷移期において高く発現していることが観察された.そこで,Mdm2の発現は細胞周期の制御にかかわるSCF複合体によって制御をうけていることを推定し,細胞周期の分野でもっとも解析が進んでいるいくつかのFボックスタンパク質を選定してそれらを細胞内でノックダウンし,Mdm2の発現に及ぼす影響を調べた.その結果,β-TRCPとよばれるFボックスタンパク質のノックダウン細胞においてのみ,特異的にMdm2の細胞内での集積がみられ,さらに,その集積がMdm2の半減期の増大によるものであることを確認した.くわえて,細胞内で内在性のβ-TRCPとMdm2との分子間結合が認められ,その相互作用がDNA損傷により増強されるという興味深いデータも得られた.これらのことから,Fボックスタンパク質としてβ-TRCPをもつSCF複合体であるSCFβ-TRCPが,Mdm2の安定性の制御に直接的にかかわっているものと結論づけた.
SCFβ-TRCPはその基質タンパク質の分解を介して,WntシグナルやNF-κBシグナル,細胞周期の制御にかかわっているほか,近年,さまざまな膜受容体や核内タンパク質を含めた幅広いシグナル伝達タンパク質の安定性を制御していることが報告されており,その多機能性が認識されるにいたっている7).SCFβ-TRCPは基質の安定性制御領域にあるDSGxxSモチーフを認識するが,基質への結合にはDSGxxSモチーフの2箇所のセリン残基のリン酸化が必要である5-7).すなわち,ユビキチンリガーゼと基質とのあいだの相互作用は基質側の翻訳後修飾による制御下におかれていて,SCFβ-TRCPの活性は,その標的となる基質に特異的なキナーゼに依存して制御されているといってよい.
Mdm2の分解制御に関与するキナーゼを特定するため,まず,PEST Finderプログラムを用いたMdm2の不安定化領域の推定から,Mdm2に存在する2箇所のPEST配列を同定した.PEST配列は自らの分解を促進させるモチーフとして知られる配列だが,Mdm2のPEST配列にはカゼインキナーゼIおよびカゼインキナーゼIIのリン酸化モチーフがクラスター化されていた.これらの予測をもとにデザインしたin vitroでの結合実験から,カゼインキナーゼIでリン酸化処理したMdm2に対してのみ,β-TRCPが特異的に結合するという結果が得られた.カゼインキナーゼIを細胞内に過剰発現させると内在性Mdm2の発現が低下する一方,カゼインキナーゼIを不活性化することによりMdm2の発現は有意に上昇したことから,カゼインキナーゼIの活性がMdm2の安定性制御に強い影響をあたえていることが確認された.カゼインキナーゼIファミリーのなかでも,カゼインキナーゼIδがMdm2の安定性制御に強く関与していることを示唆するデータも得られた.
つぎに,DNA損傷によるMdm2のすみやかな分解が,SCFβ-TRCPとカゼインキナーゼIδによってどのように誘導されているのかを解析した.これまでに,DNA損傷とβ-TRCPの機能との関連については報告されているが7),DNA損傷とカゼインキナーゼIδの活性化とを結びつける知見は乏しい.しかし近年,カゼインキナーゼIδがDNA損傷によってその局在を細胞質から核内に移行させるという興味深い報告がなされ8),筆者らの解析においても,U2OS細胞においてDNA損傷ののちにカゼインキナーゼIδとMdm2との核内での共局在が誘導されたほか,カゼインキナーゼIδとMdm2とのあいだの分子間結合もDNA損傷刺激により増強された.以上の結果から,これらカゼインキナーゼIδの挙動が,Mdm2の核内リン酸化と,それにつづくβ-TRCPのMdm2への結合を誘導し,Mdm2をSCFβ-TRCP依存的な分解へ導くという分子機構が推定された.(図1a)
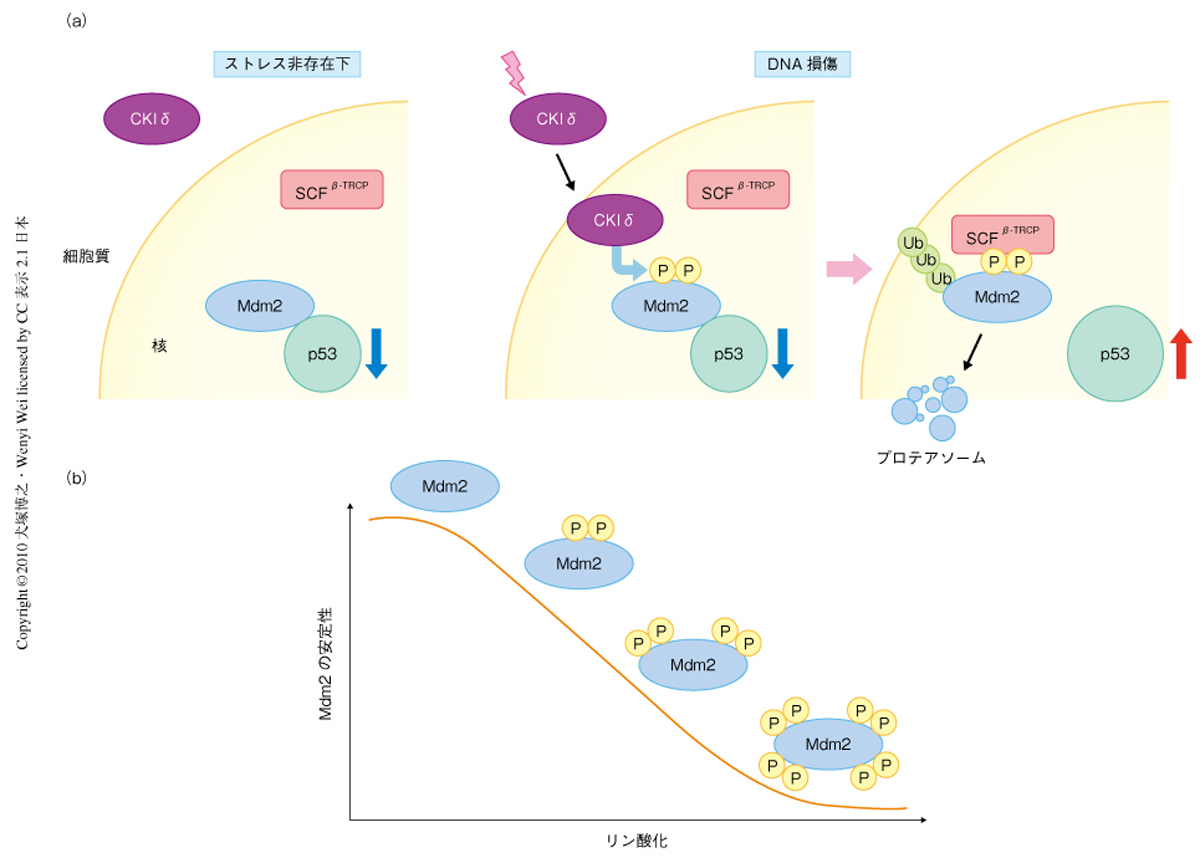
SCFβ-TRCPがMdm2を基質として認識し直接的にユビキチン化を行うユビキチンリガーゼであると結論づけるためには,β-TRCPが認識する制御領域の同定と,その制御領域への変異導入によるMdm2のカゼインキナーゼIδ-SCFβ-TRCP経路に対する分解耐性化を示す必要がある.Mdm2に存在する2箇所のPEST配列を欠失させることにより,カゼインキナーゼIδによるMdm2のリン酸化,および,β-TRCPとの相互作用はほぼ完全に消失した.よって,PEST配列に存在するリン酸化制御領域を同定するため,質量分析によってMdm2のPEST配列においてリン酸化をうけたセリン残基およびスレオニン残基を網羅的に解析した.同定されたリン酸化残基のなかから,β-TRCP認識モチーフに類似する配列に存在するセリン残基およびスレオニン残基を選び出して順にアラニン残基に変換していったところ,変異の導入数に相関して,in vitroにおけるカゼインキナーゼIによるリン酸化,および,β-TRCPのMdm2に対する結合量が減弱・消失していった.
さらに興味深いことに,細胞内でのこれら変異体の発現量は,in vitroでのカゼインキナーゼIによるリン酸化,および,β-TRCPとの結合量と,明確な逆相関を示すかたちで増加した(図1b).もっとも安定化された変異体ではその半減期が有意に増大し,かつ,SCFβ-TRCPとカゼインキナーゼIδによるin vitroでのユビキチン化をうけなかった.以上の解析結果は,Mdm2の分解が単純なオン/オフのスイッチによる1段階制御ではなく,複数のリン酸化制御領域により制御された多重制御であることを示すもので,SCFβ-TRCPによるMdm2の発現制御機構が,当初,予想していた以上に複雑で精密に制御されたものであることが明らかとなった.
p53の活性は,おもに翻訳後修飾のほか,その発現レベルによって制御されているが,多くの細胞でp53の発現は負の制御因子であるMdm2の発現と負の相関を示している.そこで,さまざまな細胞においてβ-TRCPをノックダウンすることにより,SCFβ-TRCP依存的なMdm2の発現量の制御がどのようにp53の活性化の制御に影響を及ぼしているかを調べ,以下の結果を得た.1)DNA損傷によって誘導されるMdm2のダウンレギュレーションはβ-TRCPノックダウンにより有意に抑制された.2)β-TRCPをノックダウンしたU2OS細胞での細胞周期に依存的なMdm2発現の変動において,S期初期でのMdm2の発現は対照細胞に比べ上昇しており,それと逆相関して,同じ時期のp53とその標的タンパク質p21およびBaxの発現は有意に低下していた.3)さらにこの条件下の細胞にDNA損傷刺激をあたえたところ,β-TRCPノックダウン細胞においてp53に依存的なアポトーシスが抑制された.以上の結果から,β-TRCPがMdm2の発現制御を介してp53活性を制御していることが確認された.
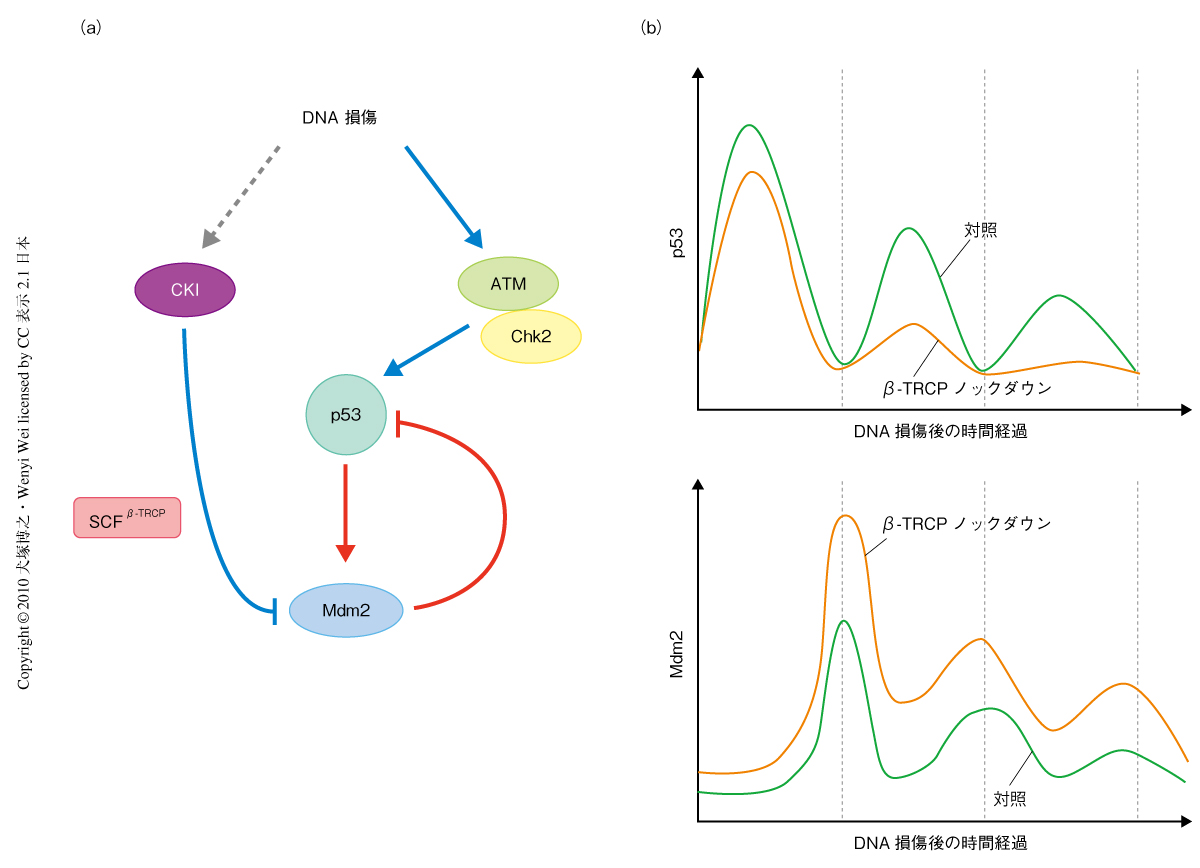
DNA損傷刺激ののちのp53の発現は,ATMおよびChk2の振動を示す活性化によるp53リン酸化(正の制御)と,p53によるMdm2遺伝子の転写活性化(負の制御)との相互の組合せによる振動を示すことが報告されている9)(図2a).β-TRCPノックダウンMCF7細胞におけるDNA損傷ののちのp53発現の振動は,第1波目は対照細胞とほぼ同じ振幅であったが,β-TRCPのノックダウンによってMdm2発現の振動においてその第1波目の振幅が増大し,このことがp53発現の振動において第2波目の振幅を大きく抑制し,さらに,そのあとにつづく振動を消失させた(図2b).このことは,SCFβ-TRCPがDNA損傷により展開されるMdm2-p53系のダイナミクスを直接的に制御していることを証明している.
ここではおもに,DNA損傷により誘導されるMdm2のユビキチン化の分子基盤に焦点をあてた.この研究において,カゼインキナーゼIとSCFβ-TRCPユビキチンリガーゼ複合体との協調作業による精密でダイナミックなMdm2分解モデルが示され,さらに,カゼインキナーゼIとSCFβ-TRCPとを介したMdm2の分解がp53の動的な挙動を直接的に制御していることが明らかとなった.このことは,ゲノムの恒常性維持にSCFβ-TRCPが重要な役割を担っていることを示している.
Mdm2はがん遺伝子産物として多くのヒト腫瘍でその過剰発現が報告されているが10),遺伝子増幅を除くMdm2高発現腫瘍において,その原因となる分子機構の多くは未解明である.この解析により得られた結果は,カゼインキナーゼIδ-SCFβ-TRCP経路を介するMdm2分解制御機構の異常が,Mdm2の発現上昇とp53の活性低下,ひいては,細胞のがん化につながるという可能性を示唆している.カゼインキナーゼIδやSCFβ-TRCPの生理機能と発がんとの直接的な関連について臨床分野における確証されたデータは少なく,今後のさらなる研究の進展が期待される.
略歴:2000年 東京大学大学院農学生命科学研究科博士課程 修了,同年 千葉県がんセンター 研修生,2001年 香川医科大学医学部 教務職員,2004年 米国Emory大学School of Medicine 研究員,2006年 米国Harvard大学Medical School 研究員を経て,2010年より同 インストラクター.
研究テーマ:SCFユビキチンリガーゼ複合体の生理機能と発がん.
抱負:ユビキチン-プロテアソーム系による多彩な生命現象を明らかにしていくことにより,タンパク質分解経路の異常によって発症する疾患の分子機序の解明,および,その治療法の開発にたずさわっていきたい.
Wenyi Wei
米国Harvard大学Medical School Assistant Professor.
© 2010 犬塚博之・Wenyi Wei Licensed under CC 表示 2.1 日本
(米国Harvard大学Medical School,Beth Israel Deaconess Medical Center,Department of Pathology)
email:犬塚博之
DOI: 10.7875/first.author.2010.018
Phosphorylation by casein kinase I promotes the turnover of the Mdm2 oncoprotein via the SCFβ-TRCP ubiquitin ligase.
Hiroyuki Inuzuka, Alan Tseng, Daming Gao, Bo Zhai, Qing Zhang, Shavali Shaik, Lixin Wan, Xiaolu L. Ang, Caroline Mock, Haoqiang Yin, Jayne M. Stommel, Steven Gygi, Galit Lahav, John Asara, Zhi-Xiong Jim Xiao, William G. Kaelin, J. Wade Harper, Wenyi Wei
Cancer Cell, 18, 147-159 (2010)
要 約
Mdm2はユビキチンリガーゼとしてがん抑制遺伝子産物p53の量的な制御をつかさどる重要なタンパク質であり,p53の活性はMdm2の発現量に応じた負の制御をうけている.Mdm2自体もユビキチン-プロテアソーム系による分解を介する発現制御をうけることが示されているが,その分子機構に関する知見は少ない.今回,筆者らは,DNA損傷により誘導されるすみやかなMdm2の分解が,カゼインキナーゼIによるMdm2のリン酸化と,それにつづくSCFβ-TRCPユビキチンリガーゼのMdm2への結合,そして,ユビキチン化,という過程により制御されていることを見い出した.さらに,SCFβ-TRCPによるMdm2の分解制御が,DNA損傷の際に出現するp53発現量の振動を直接的に制御していることも明らかにした.今回の報告は,カゼインキナーゼI-SCFβ-TRCP経路を介するMdm2の分解経路の破綻が発がんリスクを高める可能性を示唆するものであり,がんの分野の中心課題であるp53シグナル解析に新たな知見をもたらすものと期待される.
はじめに
多くのヒト腫瘍においては,がん遺伝子の活性化に付随して起こる主要ながん抑制遺伝子の機能欠損が細胞のがん化をひき起こしている.p53はもっとも重要ながん抑制遺伝子産物のひとつであり,ヒト腫瘍の半数近くにおいて変異が報告されている1).細胞がDNA損傷などのストレスを感知すると,p53はすみやかに,細胞周期の停止やアポトーシス,DNA修復などに関連する遺伝子の発現を誘導して,細胞のがん化を防いでいる.その一方で,p53の異常な活性化は予期せぬアポトーシスや細胞老化をまねくことから,p53の活性制御には非常に緻密で幾重もの制御機構が存在する.
p53のおもな制御機構としては,翻訳後修飾のほか,その量的な制御がある.ストレス非存在下の細胞においてp53の発現量は基底レベルに抑えられているが,その制御はおもにユビキチンリガーゼであるMdm2が担っている2).Mdm2はp53に結合してそのユビキチン化を行い,プロテアソームによるp53の分解を促進している.そして,いったん細胞がDNA損傷シグナルを感知すると,Mdm2はすみやかに発現量を減少・消失させることによってp53を安定化させ,p53の活性を上昇させる.したがって,Mdm2の分解制御機構はp53活性化を直接的に制御するうえで非常に重要であるが,Mdm2分解の分子機序については未解明な点が多い.当初の知見では,自己ユビキチン化がその主要な分子機構であると考えられていたが3),近年,自己ユビキチン化能を消失させたMdm2変異体を発現するノックインマウスにおいても,依然として,このMdm2変異体がユビキチン-プロテアソーム系を介して分解をうけていることが明らかとなった4).このことは,Mdm2が自己ユビキチン化以外に,未同定のユビキチンリガーゼによってユビキチン化をうけ,その発現量を変動させていることを示唆している.
このような背景のもと,この研究では,Mdm2の分解制御機構の分子基盤の解明のため,Mdm2のユビキチン化を促進するタンパク質の同定と,それらのp53活性化への関与を分子レベルで明らかにすることを主要な目的とした.
1.SCFβ-TRCPによるMdm2の安定性の制御
現在,報告もしくは存在が推定されているユビキチンリガーゼのなかでCullin-RINGフィンガー型ユビキチンリガーゼが最大のファミリーを形成しており,その数は数百にも及ぶと推定されている5).このファミリーに属するユビキチンリガーゼがMdm2の分解をつかさどっている可能性を考え,最初に,免疫沈降法により細胞内でのCullinとMdm2との相互作用の検出を試みた.そして,個々のCullinファミリータンパク質の強制発現において,Mdm2がそのファミリーのひとつCullin1とのみ特異的に相互作用することを見い出した.Cullin1はユビキチンリガーゼ複合体のなかで足場タンパク質として機能し,現在,70種類以上も報告されているFボックスタンパク質とよばれる可変的な基質認識サブユニットと,アダプタータンパク質Skp1を介して選択的に結合することで,基質認識の多様性を生み出している.これら,Skp1,Cullin1,Fボックスタンパク質からなるユビキチンリガーゼ複合体はSCF(Skp1-Cullin1-F-box protein)複合体とよばれている6).
細胞周期におけるMdm2の発現解析から,Mdm2はG1/S遷移期において高く発現していることが観察された.そこで,Mdm2の発現は細胞周期の制御にかかわるSCF複合体によって制御をうけていることを推定し,細胞周期の分野でもっとも解析が進んでいるいくつかのFボックスタンパク質を選定してそれらを細胞内でノックダウンし,Mdm2の発現に及ぼす影響を調べた.その結果,β-TRCPとよばれるFボックスタンパク質のノックダウン細胞においてのみ,特異的にMdm2の細胞内での集積がみられ,さらに,その集積がMdm2の半減期の増大によるものであることを確認した.くわえて,細胞内で内在性のβ-TRCPとMdm2との分子間結合が認められ,その相互作用がDNA損傷により増強されるという興味深いデータも得られた.これらのことから,Fボックスタンパク質としてβ-TRCPをもつSCF複合体であるSCFβ-TRCPが,Mdm2の安定性の制御に直接的にかかわっているものと結論づけた.
2.カゼインキナーゼIによるMdm2リン酸化とその安定性の制御
SCFβ-TRCPはその基質タンパク質の分解を介して,WntシグナルやNF-κBシグナル,細胞周期の制御にかかわっているほか,近年,さまざまな膜受容体や核内タンパク質を含めた幅広いシグナル伝達タンパク質の安定性を制御していることが報告されており,その多機能性が認識されるにいたっている7).SCFβ-TRCPは基質の安定性制御領域にあるDSGxxSモチーフを認識するが,基質への結合にはDSGxxSモチーフの2箇所のセリン残基のリン酸化が必要である5-7).すなわち,ユビキチンリガーゼと基質とのあいだの相互作用は基質側の翻訳後修飾による制御下におかれていて,SCFβ-TRCPの活性は,その標的となる基質に特異的なキナーゼに依存して制御されているといってよい.
Mdm2の分解制御に関与するキナーゼを特定するため,まず,PEST Finderプログラムを用いたMdm2の不安定化領域の推定から,Mdm2に存在する2箇所のPEST配列を同定した.PEST配列は自らの分解を促進させるモチーフとして知られる配列だが,Mdm2のPEST配列にはカゼインキナーゼIおよびカゼインキナーゼIIのリン酸化モチーフがクラスター化されていた.これらの予測をもとにデザインしたin vitroでの結合実験から,カゼインキナーゼIでリン酸化処理したMdm2に対してのみ,β-TRCPが特異的に結合するという結果が得られた.カゼインキナーゼIを細胞内に過剰発現させると内在性Mdm2の発現が低下する一方,カゼインキナーゼIを不活性化することによりMdm2の発現は有意に上昇したことから,カゼインキナーゼIの活性がMdm2の安定性制御に強い影響をあたえていることが確認された.カゼインキナーゼIファミリーのなかでも,カゼインキナーゼIδがMdm2の安定性制御に強く関与していることを示唆するデータも得られた.
つぎに,DNA損傷によるMdm2のすみやかな分解が,SCFβ-TRCPとカゼインキナーゼIδによってどのように誘導されているのかを解析した.これまでに,DNA損傷とβ-TRCPの機能との関連については報告されているが7),DNA損傷とカゼインキナーゼIδの活性化とを結びつける知見は乏しい.しかし近年,カゼインキナーゼIδがDNA損傷によってその局在を細胞質から核内に移行させるという興味深い報告がなされ8),筆者らの解析においても,U2OS細胞においてDNA損傷ののちにカゼインキナーゼIδとMdm2との核内での共局在が誘導されたほか,カゼインキナーゼIδとMdm2とのあいだの分子間結合もDNA損傷刺激により増強された.以上の結果から,これらカゼインキナーゼIδの挙動が,Mdm2の核内リン酸化と,それにつづくβ-TRCPのMdm2への結合を誘導し,Mdm2をSCFβ-TRCP依存的な分解へ導くという分子機構が推定された.(図1a)
3.カゼインキナーゼIδ-SCFβ-TRCP経路による協調的なMdm2分解制御の分子基盤
SCFβ-TRCPがMdm2を基質として認識し直接的にユビキチン化を行うユビキチンリガーゼであると結論づけるためには,β-TRCPが認識する制御領域の同定と,その制御領域への変異導入によるMdm2のカゼインキナーゼIδ-SCFβ-TRCP経路に対する分解耐性化を示す必要がある.Mdm2に存在する2箇所のPEST配列を欠失させることにより,カゼインキナーゼIδによるMdm2のリン酸化,および,β-TRCPとの相互作用はほぼ完全に消失した.よって,PEST配列に存在するリン酸化制御領域を同定するため,質量分析によってMdm2のPEST配列においてリン酸化をうけたセリン残基およびスレオニン残基を網羅的に解析した.同定されたリン酸化残基のなかから,β-TRCP認識モチーフに類似する配列に存在するセリン残基およびスレオニン残基を選び出して順にアラニン残基に変換していったところ,変異の導入数に相関して,in vitroにおけるカゼインキナーゼIによるリン酸化,および,β-TRCPのMdm2に対する結合量が減弱・消失していった.
さらに興味深いことに,細胞内でのこれら変異体の発現量は,in vitroでのカゼインキナーゼIによるリン酸化,および,β-TRCPとの結合量と,明確な逆相関を示すかたちで増加した(図1b).もっとも安定化された変異体ではその半減期が有意に増大し,かつ,SCFβ-TRCPとカゼインキナーゼIδによるin vitroでのユビキチン化をうけなかった.以上の解析結果は,Mdm2の分解が単純なオン/オフのスイッチによる1段階制御ではなく,複数のリン酸化制御領域により制御された多重制御であることを示すもので,SCFβ-TRCPによるMdm2の発現制御機構が,当初,予想していた以上に複雑で精密に制御されたものであることが明らかとなった.
4.カゼインキナーゼIδ-SCFβ-TRCP経路を介したp53活性化の制御機構
p53の活性は,おもに翻訳後修飾のほか,その発現レベルによって制御されているが,多くの細胞でp53の発現は負の制御因子であるMdm2の発現と負の相関を示している.そこで,さまざまな細胞においてβ-TRCPをノックダウンすることにより,SCFβ-TRCP依存的なMdm2の発現量の制御がどのようにp53の活性化の制御に影響を及ぼしているかを調べ,以下の結果を得た.1)DNA損傷によって誘導されるMdm2のダウンレギュレーションはβ-TRCPノックダウンにより有意に抑制された.2)β-TRCPをノックダウンしたU2OS細胞での細胞周期に依存的なMdm2発現の変動において,S期初期でのMdm2の発現は対照細胞に比べ上昇しており,それと逆相関して,同じ時期のp53とその標的タンパク質p21およびBaxの発現は有意に低下していた.3)さらにこの条件下の細胞にDNA損傷刺激をあたえたところ,β-TRCPノックダウン細胞においてp53に依存的なアポトーシスが抑制された.以上の結果から,β-TRCPがMdm2の発現制御を介してp53活性を制御していることが確認された.
DNA損傷刺激ののちのp53の発現は,ATMおよびChk2の振動を示す活性化によるp53リン酸化(正の制御)と,p53によるMdm2遺伝子の転写活性化(負の制御)との相互の組合せによる振動を示すことが報告されている9)(図2a).β-TRCPノックダウンMCF7細胞におけるDNA損傷ののちのp53発現の振動は,第1波目は対照細胞とほぼ同じ振幅であったが,β-TRCPのノックダウンによってMdm2発現の振動においてその第1波目の振幅が増大し,このことがp53発現の振動において第2波目の振幅を大きく抑制し,さらに,そのあとにつづく振動を消失させた(図2b).このことは,SCFβ-TRCPがDNA損傷により展開されるMdm2-p53系のダイナミクスを直接的に制御していることを証明している.
おわりに
ここではおもに,DNA損傷により誘導されるMdm2のユビキチン化の分子基盤に焦点をあてた.この研究において,カゼインキナーゼIとSCFβ-TRCPユビキチンリガーゼ複合体との協調作業による精密でダイナミックなMdm2分解モデルが示され,さらに,カゼインキナーゼIとSCFβ-TRCPとを介したMdm2の分解がp53の動的な挙動を直接的に制御していることが明らかとなった.このことは,ゲノムの恒常性維持にSCFβ-TRCPが重要な役割を担っていることを示している.
Mdm2はがん遺伝子産物として多くのヒト腫瘍でその過剰発現が報告されているが10),遺伝子増幅を除くMdm2高発現腫瘍において,その原因となる分子機構の多くは未解明である.この解析により得られた結果は,カゼインキナーゼIδ-SCFβ-TRCP経路を介するMdm2分解制御機構の異常が,Mdm2の発現上昇とp53の活性低下,ひいては,細胞のがん化につながるという可能性を示唆している.カゼインキナーゼIδやSCFβ-TRCPの生理機能と発がんとの直接的な関連について臨床分野における確証されたデータは少なく,今後のさらなる研究の進展が期待される.
文 献
- Hollstein, M., Sidransky, D., Vogelstein, B. et al.: p53 mutations in human cancers. Science, 253, 49-53 (1991)[PubMed]
- Haupt, Y., Maya, R., Kazaz, A. et al.: Mdm2 promotes the rapid degradation of p53. Nature, 387, 396-299 (1997)[PubMed]
- Stommel, J. M. & Wahl, G. M.: Accelerated MDM2 auto-degradation induced by DNA damage kinases is required for p53 activation. EMBO J., 23, 1547-1556 (2004)[PubMed]
- Itahana, K., Mao, H., Jin, A. et al.: Targeted inactivation of Mdm2 RING finger E3 ubiquitin ligase activity in the mouse reveals mechanistic insights into p53 regulation. Cancer Cell, 12, 355-366 (2007)[PubMed]
- Petroski, M. D. & Deshaies, R. J.: Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol., 6, 9-20 (2005)[PubMed]
- Cardozo, T. & Pagano, M.: The SCF ubiquitin ligase: insights into a molecular machine. Nat. Rev. Mol. Cell Biol., 5, 739-751 (2004)[PubMed]
- Nakayama, K. I. & Nakayama, K.: Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer, 6, 369-381 (2006)[PubMed]
- Alsheich-Bartok, O., Haupt, S., Alkalay-Snir, I. et al.: PML enhances the regulation of p53 by CK1 in response to DNA damage. Oncogene, 27, 3653-3661 (2008)[PubMed]
- Batchelor, E., Mock, C. S., Bhan, I. et al.: Recurrent initiation: a mechanism for triggering p53 pulses in response to DNA damage. Mol. Cell, 30, 277-289 (2008)[PubMed]
- Rayburn, E., Zhang, R., He, J. et al.: MDM2 and human malignancies: expression, clinical pathology, prognostic markers, and implications for chemotherapy. Curr. Cancer Drug Targets, 5, 27-41 (2005)[PubMed]
著者プロフィール
略歴:2000年 東京大学大学院農学生命科学研究科博士課程 修了,同年 千葉県がんセンター 研修生,2001年 香川医科大学医学部 教務職員,2004年 米国Emory大学School of Medicine 研究員,2006年 米国Harvard大学Medical School 研究員を経て,2010年より同 インストラクター.
研究テーマ:SCFユビキチンリガーゼ複合体の生理機能と発がん.
抱負:ユビキチン-プロテアソーム系による多彩な生命現象を明らかにしていくことにより,タンパク質分解経路の異常によって発症する疾患の分子機序の解明,および,その治療法の開発にたずさわっていきたい.
Wenyi Wei
米国Harvard大学Medical School Assistant Professor.
© 2010 犬塚博之・Wenyi Wei Licensed under CC 表示 2.1 日本
