転写因子IRF8の神経炎症における役割
吉見竜介・尾里啓子
(米国NIH National Institute of Child Health and Human Development,Program in Genomics of Differentiation)
email:吉見竜介,尾里啓子
DOI: 10.7875/first.author.2014.029
The transcription factor IRF8 activates integrin-mediated TGF-β signaling and promotes neuroinflammation.
Yuko Yoshida, Ryusuke Yoshimi, Hiroaki Yoshii, Daniel Kim, Anup Dey, Huabao Xiong, Jeeva Munasinghe, Itaru Yazawa, Michael J. O’Donovan, Olga A. Maximova, Suveena Sharma, Jinfang Zhu, Hongsheng Wang, Herbert C. Morse, Keiko Ozato
Immunity, 40, 187-198 (2014)
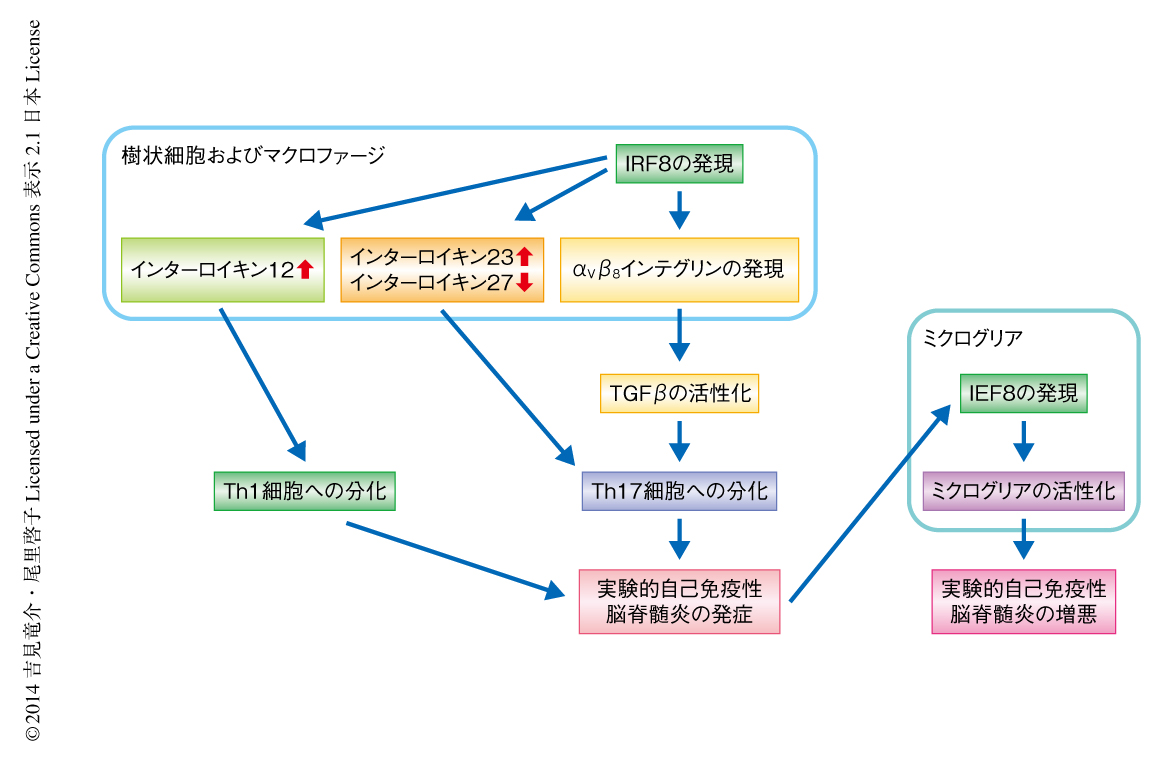
最近の疫学研究により,多発性硬化症における疾患感受性遺伝子のひとつとして転写因子IRF8をコードする遺伝子が同定されたが,この遺伝子が病態にどのような役割をもつのかについては明らかにされていなかった.今回,筆者らは,多発性硬化症のモデルである実験的自己免疫性脳脊髄炎を発症させたマウスを用いてIRF8の役割について検討した.その結果,IRF8ノックアウトマウスは実験的自己免疫性脳脊髄炎の発症に対し抵抗性を示し,抗原提示細胞にて発現するIRF8がその発症において,以下の3つの点で重要であることを見い出した.すなわち,IRF8は抗原提示細胞におけるαVβ8インテグリンの発現を亢進することによりTGFβシグナル伝達系を活性化しTh17細胞への分化を誘導する.IRF8は実験的自己免疫性脳脊髄炎の発症においてインターロイキン12およびインターロイキン23の産生を促進しインターロイキン27の産生を抑制することによりTh1細胞およびTh17細胞への分化と維持に適したサイトカインの環境を形成する.IRF8はミクログリアを活性化することにより神経炎症を増悪させる.以上の結果から,多発性硬化症の病態におけるIRF8の重要性が示唆された.
Th17細胞は炎症を促進する作用や組織に障害をおよぼす作用をもち,多くの自己免疫疾患との関連性が報告されている.ナイーブT細胞はTGFβやインターロイキン6などのサイトカインの存在のもとで,樹状細胞やマクロファージなどの抗原提示細胞のはたらきによりTh17細胞へと分化する.最近の研究により,抗原提示細胞に存在するインテグリンがTGFβシグナル伝達系を介し活性型TGFβをナイーブT細胞へと直接に供給することが明らかになってきた1,2).TGFβシグナル伝達系により,Th17細胞への分化を指揮するRORファミリー転写因子が活性化する.
Th17細胞の関連する自己免疫疾患のモデルとしてもっともよく研究されているもののひとつに,多発性硬化症のモデルである実験的自己免疫性脳脊髄炎を発症させたマウスがある.多発性硬化症は脱髄およびニューロンの損傷を起こす中枢神経系の炎症性の疾患であり,欧米の白人に多く北欧では有病率は人口10万人に50~100人,わが国では有病率は低く人口10万人あたり8~9人と推定されている.RORα,RORγt,あるいは,インターロイキン17を欠損したマウスは実験的自己免疫性脳脊髄炎の発症に対し抵抗性を示す3,4).これらのマウスと同様に,多発性硬化症を発症した患者の中枢神経系においてもTh17細胞およびインターロイキン17の存在が認められている5).
多発性硬化症の病因についてはこれまでほとんど明らかにされていなかったが,近年のゲノムワイドSNP解析により疾患感受性遺伝子が同定され,病態に対する理解への寄与が期待されている.そのなかで,樹状細胞やマクロファージの分化を制御するIRF(interferon regulatory factor)ファミリー転写因子のひとつ,IRF8をコードする遺伝子が同定された6).IRF8は樹状細胞やマクロファージにおいてインターロイキン12 p40サブユニットやI型インターフェロンをコードする遺伝子の転写を誘導することにより,さまざまな病原体に対する防御において重要な役割を担っているほか,T細胞やB細胞の活性を制御している7,8).多発性硬化症の疾患感受性と関連するSNPはIRF8遺伝子の3’側非コード領域にあり,疾患感受性の原因としてはIRF8遺伝子の転写制御における異常が考えられた.しかし,これまでIRF8が多発性硬化症にあたえる影響を示す情報はほとんどなかった.
今回,筆者らは,IRF8ノックアウトマウスが実験的自己免疫性脳脊髄炎の発症に対し抵抗性を示し,抗原提示細胞において発現したIRF8がエフェクターT細胞への分化やミクログリアによる神経炎症を促進することにより疾患をひき起こすことを明らかにした.
実験的自己免疫性脳脊髄炎の発症機序におけるIRF8の役割について調べるため,野生型マウスおよびIRF8ノックアウトマウスに自己抗原を投与し,実験的自己免疫性脳脊髄炎による運動麻痺の重症度を経時的に観察した.すべての野生型マウスにおいて自己抗原の投与ののち7~9日より運動麻痺が出現し17~20日にピークをむかえたのに対し,IRF8ノックアウトマウスでは実験的自己免疫性脳脊髄炎の発症はまったくみられなかった.投与ののち21日における脊髄組織の所見では,野生型マウスには単核球の浸潤,ニューロンの傷害,脱髄といった実験的自己免疫性脳脊髄炎に典型的な所見がみられたが,IRF8ノックアウトマウスは正常であった.MRIによる検査でも,野生型マウスでは脊髄に病変を認めたのに対し,IRF8ノックアウトマウスには異常を認めなかった.以上から,野生型マウスにおいては実験的自己免疫性脳脊髄炎が発症するような条件においても,IRF8ノックアウトマウスは実験的自己免疫性脳脊髄炎を発症しないことがわかった.
自己抗原による免疫は自己反応性のTh1細胞,Th17細胞,制御性T細胞への分化を誘導することが知られている9,10).そこで,IRF8ノックアウトマウスに自己抗原を投与したのち,Th1細胞およびTh17細胞に特徴的なサイトカインであるインターロイキン17Aおよびインターフェロンγ,および,制御性T細胞に特異的な転写因子であるFoxp3をそれぞれコードするmRNAの量を測定した.野生型マウスのリンパ節,脾臓,脊髄においては自己抗原の投与によりこれらのmRNAの増加を認めたのに対し,IRF8ノックアウトマウスではほとんど検出されなかった.IRF8ノックアウトマウスの臓器におけるTh1細胞,Th17細胞,制御性T細胞の割合をフローサイトメトリーを用いて調べたところ,いずれも野生型マウスと比較していちじるしく低下していた.自己抗原による免疫から2日のちに脊髄およびリンパ節から単核球を分離し,in vitroにおいて自己抗原による再刺激をくわえたところ,野生型マウスに由来する単核球においては抗原に特異的なTh1細胞,Th17細胞,制御性T細胞が増加したが,IRF8ノックアウトマウスに由来する単核球においてはこれらの細胞は増加しなかった.以上の結果から,IRF8ノックアウトマウスは自己抗原に反応したナイーブT細胞からエフェクターT細胞への分化を誘導できないことが示された.
IRF8ノックアウトマウスにおいてエフェクターT細胞が分化しない原因として,2つの理由が考えられた.すなわち,IRF8を欠損したT細胞においてTh1細胞あるいはTh17細胞への分化能が欠損している可能性と,IRF8を欠損した抗原提示細胞においてエフェクターT細胞の産生能が欠損している可能性である.そこで,どの細胞に発現するIRF8が実験的自己免疫性脳脊髄炎の発症に影響をあたえるのかを調べるため,IRF8のコンディショナルノックアウトマウスを用いた.その結果,単球およびマクロファージに特異的なIRF8ノックアウトマウスは実験的自己免疫性脳脊髄炎の発症に対し抵抗性を示したのに対し,T細胞に特異的なIRF8ノックアウトマウスは野生型マウスと同等の神経症状を示したことから,単球およびマクロファージに発現したIRF8が実験的自己免疫性脳脊髄炎の発症に一義的な責任をもつことが明らかになった.また,IRF8ノックアウトマウスに野生型マウスに由来する樹状細胞を移植すると,自己抗原の投与ののち13日までは実験的自己免疫性脳脊髄炎の症状を示したことから,樹状細胞に発現したIRF8も実験的自己免疫性脳脊髄炎の発症を促進する作用をもつことが裏づけられた.
実験的自己免疫性脳脊髄炎の発症においてIRF8を発現する細胞を同定するため,内在性のIRF8遺伝子プロモーターによりIRF8-GFP融合タンパク質を発現するトランスジェニックマウスから組織を取り出し,その細胞をフローサイトメトリーを用いて解析した.自己抗原による免疫のまえの脾臓におけるIRF8-GFP融合タンパク質の発現は樹状細胞およびマクロファージにおいて高く,B細胞ではやや低く,T細胞および顆粒球においてはほとんどみられなかった.また,脊髄においてIRF8-GFP融合タンパク質はマクロファージとは異なる細胞集団であるミクログリアにも発現していた.このトランスジェニックマウスを自己抗原により免疫したところ,ホモ接合型およびヘテロ接合型とも,野生型マウスと同等の神経症状を示した.実験的自己免疫性脳脊髄炎の発症においてはミクログリアにおけるIRF8-GFP融合タンパク質の発現が上昇するとともに,IRF8-GFP融合タンパク質を発現するミクログリアの数もいちじるしく増加した.脊髄においてIRF8-GFP融合タンパク質を発現するマクロファージの数も増加していたが,ミクログリアよりは少数であった.自己抗原による免疫のまえのリンパ節および脾臓においては形質細胞様樹状細胞および骨髄系樹状細胞に相当する2つの樹状細胞サブセットを認めたが,実験的自己免疫性脳脊髄炎の発症ののちにはGFPの蛍光強度が増加したとともに,形質細胞様樹状細胞に相当する樹状細胞サブセットの細胞数が増加した.実験的自己免疫性脳脊髄炎の発症においてはリンパ節や脾臓のマクロファージでもIRF8-GFP融合タンパク質の発現は上昇していた.樹状細胞およびマクロファージの数は自己抗原の投与ののち7~14日でいちじるしく増加しており,これらの大多数はIRF8-GFP融合タンパク質を発現していた.
インターロイキン12ファミリーに属するインターロイキン12,インターロイキン23,インターロイキン27は抗原提示細胞において産生され,実験的自己免疫性脳脊髄炎の発症に影響をあたえることが知られている11).インターロイキン23はインターロイキン12 p40サブユニットおよびインターロイキン23 p19サブユニットから構成されTh17細胞の増殖を促進する.インターロイキン12はp40サブユニットおよびp35サブユニットからなるヘテロ二量体でありTh1細胞への分化に関与する.インターロイキン27はp28サブユニットおよびEbi3から構成され,Th17細胞やそのほかのT細胞への分化を抑制し実験的自己免疫性脳脊髄炎の発症を抑制する.IRF8は病原体による刺激やToll様受容体への刺激によりインターロイキン12 p40サブユニットをコードする遺伝子の転写活性を亢進することから,IRF8がこれらのサイトカインの産生を制御することにより実験的自己免疫性脳脊髄炎の発症に寄与している可能性が考えられた.
おのおののサイトカインをコードするmRNAの発現について調べたところ,インターロイキン12 p40サブユニットの発現は野生型マウスではいちじるしく上昇したのに対し,IRF8ノックアウトマウスでは上昇しなかった.血清におけるインターロイキン12 p40サブユニットの発現も,野生型マウスでは実験的自己免疫性脳脊髄炎の発症においていちじるしく上昇したが,IRF8ノックアウトマウスでは上昇しなかった.インターロイキン12およびインターロイキン23の生物学的な活性はインターロイキン12 p40サブユニットに依存していることから,IRF8を欠損した抗原提示細胞は実験的自己免疫性脳脊髄炎の発症をひき起こせるようなサイトカインの環境を形成できないと考えられた.Ebi3をコードするmRNAの発現も実験的自己免疫性脳脊髄炎の発症において上昇したが,IRF8ノックアウトマウスにおける発現のほうが野生型マウスより一貫して高かった.さらに,実験的自己免疫性脳脊髄炎を発症している野生型マウスと比較して,IRF8ノックアウトマウスではインターロイキン27を産生するマクロファージやミクログリアの数が多く,血清におけるインターロイキン27の濃度についても野生型マウスよりIRF8ノックアウトマウスのほうが高かった.IRF8ノックアウトマウスにおけるインターロイキン27の産生の増加がTh17細胞への分化の抑制に寄与しているかどうかを調べるため,CD4陽性T細胞と自己抗原の投与ののち21日のIRF8ノックアウトマウスから採取した樹状細胞とをin vitroにおいて共培養したところ,抗インターロイキン27抗体の存在のもとでは野生型の樹状細胞と同じ程度のTh17細胞への分化およびインターロイキン17の発現がみられた.以上の結果から,IRF8がインターロイキン12ファミリーの発現を制御することにより,実験的自己免疫性脳脊髄炎の発症をひき起こせるようなサイトカインの環境を形成していることが示された.
ここまでの結果より,インターロイキン12ファミリーが実験的自己免疫性脳脊髄炎の発症に寄与することは示されたが,IRF8ノックアウトマウスにおいてエフェクターTh17細胞への分化がまったくみられないことについては説明できず,もっと早期の感作の段階において異常の存在する可能性が示唆された.抗原提示細胞におけるTGFβシグナル伝達系の活性化はTh17細胞への分化を開始し実験的自己免疫性脳脊髄炎が発症するために重要であり1,2),IRF8がαVβ8インテグリンをコードする遺伝子の転写を制御することによりこの段階に作用していると仮定した.IRF8ノックアウトマウスに由来するマクロファージおよび樹状細胞におけるαVβ8インテグリン遺伝子のmRNAの発現を野生型マウスと比較したところ,野生型マウスでは発現が高かったのに対しIRF8ノックアウトマウスでは発現は非常に低かった.さらに,野生型マウスの骨髄に由来する樹状細胞およびマクロファージをインターフェロンγおよびリポ多糖により刺激するとαVβ8インテグリン遺伝子の転写が誘導されたのに対し,IRF8ノックアウトマウスではその誘導は軽微であった.αVβ8インテグリン遺伝子プロモーターの断片を含むレポーターベクターおよびIRF8の発現ベクターを293T細胞株に共導入して行ったルシフェラーゼアッセイにより,IRF8がαVβ8インテグリン遺伝子プロモーターの活性を増強させ,それにはプロモーター領域の-1081~-1952の領域が必要であることが示唆された.この領域にはインターフェロン応答配列に似た複数のIRF8結合予想部位が含まれており,そのひとつに変異を導入したところレポーター活性が失われたことから,αVβ8インテグリン遺伝子の転写におけるこの部位の関与が裏づけられた.さらに,内在性のIRF8遺伝子プロモーターによりIRF8-GFP融合タンパク質を発現するトランスジェニックマウスの骨髄に由来する樹状細胞に対し,GFPおよびIRF8に対する抗体を用いてクロマチン免疫沈降(chromatin immunoprecipitation:ChIP)法を行った結果,IRF8がルシフェラーゼアッセイにおいて推定された部位と結合することが確認された.以上から,IRF8がαVβ8インテグリン遺伝子の転写活性を直接に増強していることが明らかになった.
IRF8が抗原提示細胞によるTGFβシグナル伝達系の活性化を介してナイーブT細胞のエフェクターT細胞への分化に関与しているのかどうかを調べるため,CD11c陽性の樹状細胞とナイーブT細胞をin vitroにおいて生物学的な活性をもたない潜在型TGFβの存在のもとで共培養した.その結果,野生型の樹状細胞はTh17細胞への分化を促進したが,IRF8を欠損した樹状細胞は促進しなかった.一方,活性型TGFβの存在のもとでは野生型の樹状細胞およびIRF8を欠損した樹状細胞はともにTh17細胞へと分化したことから,IRF8を欠損した樹状細胞は潜在型TGFβを活性化することのできないことが示唆された.IRF8を欠損した樹状細胞の培養上清に含まれるインターロイキン17の量についても,潜在型TGFβの存在のもとでは低値であったが活性型TGFβの存在のもとでは野生型の樹状細胞と同等であった.さらに,IRF8を欠損した樹状細胞は潜在型TGFβの存在のもとでは制御性T細胞への分化を促進しなかったのに対し,野生型の樹状細胞はこれを促進したことから,IRF8を欠損した樹状細胞においてナイーブT細胞からエフェクター細胞へと分化できない原因はTGFβシグナル伝達系の欠損にあるという考えが裏づけられた.
TGFβシグナル伝達系により活性化されるSMADファミリー転写因子のうち,SMAD2はTh17細胞の活性化において重要とされている12).野生型の樹状細胞は潜在型TGFβの存在のもとでT細胞においてSMAD2のリン酸化を増強したのに対し,IRF8を欠損した樹状細胞は増強しなかった.その一方で,活性型TGFβの存在のもとでは野生型の樹状細胞およびIRF8を欠損した樹状細胞はともにSMAD2のリン酸化を増強した.in vivoにおいても,野生型マウスでは実験的自己免疫性脳脊髄炎の発症においてCD4陽性T細胞におけるSMAD2のリン酸化が増強したのに対し,IRF8ノックアウトマウスでは増強しなかった.さらに,IRF8およびαVβ8インテグリンをコードするmRNAの量が実験的自己免疫性脳脊髄炎を発症している野生型マウスの樹状細胞においては増加していたのに対し,IRF8ノックアウトマウスの樹状細胞では増加していなかった.実験的自己免疫性脳脊髄炎を発症している野生型マウスと比較して,IRF8ノックアウトマウスでは血清における活性型TGFβの濃度は非常に低値であった.これにくわえて,TGFβおよびインターロイキン6をコードするmRNAの量は実験的自己免疫性脳脊髄炎を発症している野生型マウスとIRF8ノックアウトマウスとは同等であったことから,IRF8ノックアウトマウスでは生物学的な活性をもつ活性型TGFβの産生が選択的に障害されていることが示された.
この研究により,筆者らは,多発性硬化症のモデルである実験的自己免疫性脳脊髄炎を発症したマウスにおいて,抗原提示細胞におけるαVβ8インテグリンを介したTGFβの活性化からナイーブT細胞の感作によるエフェクター細胞への分化にいたる過程にて,転写因子IRF8が多面的に必要であるという強力で新しい証拠を得た(図1).IRF8遺伝子は多発性硬化症の疾患感受性遺伝子として同定されているが,その責任SNPは3’側非コード領域に位置する.したがって,感受性の原因としてはIRF8遺伝子の転写制御における異常が考えられるが,現在のところ,このSNPがIRF8遺伝子の転写にどのような影響をあたえているのかは不明である.さらに,多発性硬化症に対する現在の治療法がIRF8の発現や機能に対しどのような影響をあたえているのか,あるいは,Th17細胞が関与するとされるほかの自己免疫疾患においても同様の作用によりIRF8が病態に影響をあたえているのかなど,明らかにすべき点は多く残されており,この分野におけるこれからの研究が期待される.
略歴:2006年 横浜市立大学大学院医学研究科博士課程 修了,同年 横浜市立大学附属病院 常勤特別職,2007年 米国NIH National Institute of Child Health and Human Developmentポストドクトラルビジティングフェローを経て,2010年より横浜市立大学大学院医学研究科 特任助教(現 助教).
研究テーマ:全身性自己免疫疾患の発症の機序と臨床への応用.
尾里 啓子(Keiko Ozato)
米国NIH National Institute of Child Health and Human Development部門主任.
研究室URL:https://science.nichd.nih.gov/confluence/display/pgd/Keiko+Ozato+Lab
© 2014 吉見竜介・尾里啓子 Licensed under CC 表示 2.1 日本
(米国NIH National Institute of Child Health and Human Development,Program in Genomics of Differentiation)
email:吉見竜介,尾里啓子
DOI: 10.7875/first.author.2014.029
The transcription factor IRF8 activates integrin-mediated TGF-β signaling and promotes neuroinflammation.
Yuko Yoshida, Ryusuke Yoshimi, Hiroaki Yoshii, Daniel Kim, Anup Dey, Huabao Xiong, Jeeva Munasinghe, Itaru Yazawa, Michael J. O’Donovan, Olga A. Maximova, Suveena Sharma, Jinfang Zhu, Hongsheng Wang, Herbert C. Morse, Keiko Ozato
Immunity, 40, 187-198 (2014)
要 約
最近の疫学研究により,多発性硬化症における疾患感受性遺伝子のひとつとして転写因子IRF8をコードする遺伝子が同定されたが,この遺伝子が病態にどのような役割をもつのかについては明らかにされていなかった.今回,筆者らは,多発性硬化症のモデルである実験的自己免疫性脳脊髄炎を発症させたマウスを用いてIRF8の役割について検討した.その結果,IRF8ノックアウトマウスは実験的自己免疫性脳脊髄炎の発症に対し抵抗性を示し,抗原提示細胞にて発現するIRF8がその発症において,以下の3つの点で重要であることを見い出した.すなわち,IRF8は抗原提示細胞におけるαVβ8インテグリンの発現を亢進することによりTGFβシグナル伝達系を活性化しTh17細胞への分化を誘導する.IRF8は実験的自己免疫性脳脊髄炎の発症においてインターロイキン12およびインターロイキン23の産生を促進しインターロイキン27の産生を抑制することによりTh1細胞およびTh17細胞への分化と維持に適したサイトカインの環境を形成する.IRF8はミクログリアを活性化することにより神経炎症を増悪させる.以上の結果から,多発性硬化症の病態におけるIRF8の重要性が示唆された.
はじめに
Th17細胞は炎症を促進する作用や組織に障害をおよぼす作用をもち,多くの自己免疫疾患との関連性が報告されている.ナイーブT細胞はTGFβやインターロイキン6などのサイトカインの存在のもとで,樹状細胞やマクロファージなどの抗原提示細胞のはたらきによりTh17細胞へと分化する.最近の研究により,抗原提示細胞に存在するインテグリンがTGFβシグナル伝達系を介し活性型TGFβをナイーブT細胞へと直接に供給することが明らかになってきた1,2).TGFβシグナル伝達系により,Th17細胞への分化を指揮するRORファミリー転写因子が活性化する.
Th17細胞の関連する自己免疫疾患のモデルとしてもっともよく研究されているもののひとつに,多発性硬化症のモデルである実験的自己免疫性脳脊髄炎を発症させたマウスがある.多発性硬化症は脱髄およびニューロンの損傷を起こす中枢神経系の炎症性の疾患であり,欧米の白人に多く北欧では有病率は人口10万人に50~100人,わが国では有病率は低く人口10万人あたり8~9人と推定されている.RORα,RORγt,あるいは,インターロイキン17を欠損したマウスは実験的自己免疫性脳脊髄炎の発症に対し抵抗性を示す3,4).これらのマウスと同様に,多発性硬化症を発症した患者の中枢神経系においてもTh17細胞およびインターロイキン17の存在が認められている5).
多発性硬化症の病因についてはこれまでほとんど明らかにされていなかったが,近年のゲノムワイドSNP解析により疾患感受性遺伝子が同定され,病態に対する理解への寄与が期待されている.そのなかで,樹状細胞やマクロファージの分化を制御するIRF(interferon regulatory factor)ファミリー転写因子のひとつ,IRF8をコードする遺伝子が同定された6).IRF8は樹状細胞やマクロファージにおいてインターロイキン12 p40サブユニットやI型インターフェロンをコードする遺伝子の転写を誘導することにより,さまざまな病原体に対する防御において重要な役割を担っているほか,T細胞やB細胞の活性を制御している7,8).多発性硬化症の疾患感受性と関連するSNPはIRF8遺伝子の3’側非コード領域にあり,疾患感受性の原因としてはIRF8遺伝子の転写制御における異常が考えられた.しかし,これまでIRF8が多発性硬化症にあたえる影響を示す情報はほとんどなかった.
今回,筆者らは,IRF8ノックアウトマウスが実験的自己免疫性脳脊髄炎の発症に対し抵抗性を示し,抗原提示細胞において発現したIRF8がエフェクターT細胞への分化やミクログリアによる神経炎症を促進することにより疾患をひき起こすことを明らかにした.
1.IRF8ノックアウトマウスは実験的自己免疫性脳脊髄炎を発症しない
実験的自己免疫性脳脊髄炎の発症機序におけるIRF8の役割について調べるため,野生型マウスおよびIRF8ノックアウトマウスに自己抗原を投与し,実験的自己免疫性脳脊髄炎による運動麻痺の重症度を経時的に観察した.すべての野生型マウスにおいて自己抗原の投与ののち7~9日より運動麻痺が出現し17~20日にピークをむかえたのに対し,IRF8ノックアウトマウスでは実験的自己免疫性脳脊髄炎の発症はまったくみられなかった.投与ののち21日における脊髄組織の所見では,野生型マウスには単核球の浸潤,ニューロンの傷害,脱髄といった実験的自己免疫性脳脊髄炎に典型的な所見がみられたが,IRF8ノックアウトマウスは正常であった.MRIによる検査でも,野生型マウスでは脊髄に病変を認めたのに対し,IRF8ノックアウトマウスには異常を認めなかった.以上から,野生型マウスにおいては実験的自己免疫性脳脊髄炎が発症するような条件においても,IRF8ノックアウトマウスは実験的自己免疫性脳脊髄炎を発症しないことがわかった.
2.IRF8ノックアウトマウスは自己抗原を投与してもエフェクターT細胞を誘導しない
自己抗原による免疫は自己反応性のTh1細胞,Th17細胞,制御性T細胞への分化を誘導することが知られている9,10).そこで,IRF8ノックアウトマウスに自己抗原を投与したのち,Th1細胞およびTh17細胞に特徴的なサイトカインであるインターロイキン17Aおよびインターフェロンγ,および,制御性T細胞に特異的な転写因子であるFoxp3をそれぞれコードするmRNAの量を測定した.野生型マウスのリンパ節,脾臓,脊髄においては自己抗原の投与によりこれらのmRNAの増加を認めたのに対し,IRF8ノックアウトマウスではほとんど検出されなかった.IRF8ノックアウトマウスの臓器におけるTh1細胞,Th17細胞,制御性T細胞の割合をフローサイトメトリーを用いて調べたところ,いずれも野生型マウスと比較していちじるしく低下していた.自己抗原による免疫から2日のちに脊髄およびリンパ節から単核球を分離し,in vitroにおいて自己抗原による再刺激をくわえたところ,野生型マウスに由来する単核球においては抗原に特異的なTh1細胞,Th17細胞,制御性T細胞が増加したが,IRF8ノックアウトマウスに由来する単核球においてはこれらの細胞は増加しなかった.以上の結果から,IRF8ノックアウトマウスは自己抗原に反応したナイーブT細胞からエフェクターT細胞への分化を誘導できないことが示された.
3.実験的自己免疫性脳脊髄炎の発症には単球およびマクロファージにおけるIRF8の発現が重要である
IRF8ノックアウトマウスにおいてエフェクターT細胞が分化しない原因として,2つの理由が考えられた.すなわち,IRF8を欠損したT細胞においてTh1細胞あるいはTh17細胞への分化能が欠損している可能性と,IRF8を欠損した抗原提示細胞においてエフェクターT細胞の産生能が欠損している可能性である.そこで,どの細胞に発現するIRF8が実験的自己免疫性脳脊髄炎の発症に影響をあたえるのかを調べるため,IRF8のコンディショナルノックアウトマウスを用いた.その結果,単球およびマクロファージに特異的なIRF8ノックアウトマウスは実験的自己免疫性脳脊髄炎の発症に対し抵抗性を示したのに対し,T細胞に特異的なIRF8ノックアウトマウスは野生型マウスと同等の神経症状を示したことから,単球およびマクロファージに発現したIRF8が実験的自己免疫性脳脊髄炎の発症に一義的な責任をもつことが明らかになった.また,IRF8ノックアウトマウスに野生型マウスに由来する樹状細胞を移植すると,自己抗原の投与ののち13日までは実験的自己免疫性脳脊髄炎の症状を示したことから,樹状細胞に発現したIRF8も実験的自己免疫性脳脊髄炎の発症を促進する作用をもつことが裏づけられた.
4.実験的自己免疫性脳脊髄炎の発症においてはマクロファージ,樹状細胞,ミクログリアにおけるIRF8の発現が亢進する
実験的自己免疫性脳脊髄炎の発症においてIRF8を発現する細胞を同定するため,内在性のIRF8遺伝子プロモーターによりIRF8-GFP融合タンパク質を発現するトランスジェニックマウスから組織を取り出し,その細胞をフローサイトメトリーを用いて解析した.自己抗原による免疫のまえの脾臓におけるIRF8-GFP融合タンパク質の発現は樹状細胞およびマクロファージにおいて高く,B細胞ではやや低く,T細胞および顆粒球においてはほとんどみられなかった.また,脊髄においてIRF8-GFP融合タンパク質はマクロファージとは異なる細胞集団であるミクログリアにも発現していた.このトランスジェニックマウスを自己抗原により免疫したところ,ホモ接合型およびヘテロ接合型とも,野生型マウスと同等の神経症状を示した.実験的自己免疫性脳脊髄炎の発症においてはミクログリアにおけるIRF8-GFP融合タンパク質の発現が上昇するとともに,IRF8-GFP融合タンパク質を発現するミクログリアの数もいちじるしく増加した.脊髄においてIRF8-GFP融合タンパク質を発現するマクロファージの数も増加していたが,ミクログリアよりは少数であった.自己抗原による免疫のまえのリンパ節および脾臓においては形質細胞様樹状細胞および骨髄系樹状細胞に相当する2つの樹状細胞サブセットを認めたが,実験的自己免疫性脳脊髄炎の発症ののちにはGFPの蛍光強度が増加したとともに,形質細胞様樹状細胞に相当する樹状細胞サブセットの細胞数が増加した.実験的自己免疫性脳脊髄炎の発症においてはリンパ節や脾臓のマクロファージでもIRF8-GFP融合タンパク質の発現は上昇していた.樹状細胞およびマクロファージの数は自己抗原の投与ののち7~14日でいちじるしく増加しており,これらの大多数はIRF8-GFP融合タンパク質を発現していた.
5.IRF8ノックアウトマウスはインターロイキン12ファミリーの産生に異常がある
インターロイキン12ファミリーに属するインターロイキン12,インターロイキン23,インターロイキン27は抗原提示細胞において産生され,実験的自己免疫性脳脊髄炎の発症に影響をあたえることが知られている11).インターロイキン23はインターロイキン12 p40サブユニットおよびインターロイキン23 p19サブユニットから構成されTh17細胞の増殖を促進する.インターロイキン12はp40サブユニットおよびp35サブユニットからなるヘテロ二量体でありTh1細胞への分化に関与する.インターロイキン27はp28サブユニットおよびEbi3から構成され,Th17細胞やそのほかのT細胞への分化を抑制し実験的自己免疫性脳脊髄炎の発症を抑制する.IRF8は病原体による刺激やToll様受容体への刺激によりインターロイキン12 p40サブユニットをコードする遺伝子の転写活性を亢進することから,IRF8がこれらのサイトカインの産生を制御することにより実験的自己免疫性脳脊髄炎の発症に寄与している可能性が考えられた.
おのおののサイトカインをコードするmRNAの発現について調べたところ,インターロイキン12 p40サブユニットの発現は野生型マウスではいちじるしく上昇したのに対し,IRF8ノックアウトマウスでは上昇しなかった.血清におけるインターロイキン12 p40サブユニットの発現も,野生型マウスでは実験的自己免疫性脳脊髄炎の発症においていちじるしく上昇したが,IRF8ノックアウトマウスでは上昇しなかった.インターロイキン12およびインターロイキン23の生物学的な活性はインターロイキン12 p40サブユニットに依存していることから,IRF8を欠損した抗原提示細胞は実験的自己免疫性脳脊髄炎の発症をひき起こせるようなサイトカインの環境を形成できないと考えられた.Ebi3をコードするmRNAの発現も実験的自己免疫性脳脊髄炎の発症において上昇したが,IRF8ノックアウトマウスにおける発現のほうが野生型マウスより一貫して高かった.さらに,実験的自己免疫性脳脊髄炎を発症している野生型マウスと比較して,IRF8ノックアウトマウスではインターロイキン27を産生するマクロファージやミクログリアの数が多く,血清におけるインターロイキン27の濃度についても野生型マウスよりIRF8ノックアウトマウスのほうが高かった.IRF8ノックアウトマウスにおけるインターロイキン27の産生の増加がTh17細胞への分化の抑制に寄与しているかどうかを調べるため,CD4陽性T細胞と自己抗原の投与ののち21日のIRF8ノックアウトマウスから採取した樹状細胞とをin vitroにおいて共培養したところ,抗インターロイキン27抗体の存在のもとでは野生型の樹状細胞と同じ程度のTh17細胞への分化およびインターロイキン17の発現がみられた.以上の結果から,IRF8がインターロイキン12ファミリーの発現を制御することにより,実験的自己免疫性脳脊髄炎の発症をひき起こせるようなサイトカインの環境を形成していることが示された.
6.IRF8は抗原提示細胞におけるαVβ8インテグリンの発現に関与する
ここまでの結果より,インターロイキン12ファミリーが実験的自己免疫性脳脊髄炎の発症に寄与することは示されたが,IRF8ノックアウトマウスにおいてエフェクターTh17細胞への分化がまったくみられないことについては説明できず,もっと早期の感作の段階において異常の存在する可能性が示唆された.抗原提示細胞におけるTGFβシグナル伝達系の活性化はTh17細胞への分化を開始し実験的自己免疫性脳脊髄炎が発症するために重要であり1,2),IRF8がαVβ8インテグリンをコードする遺伝子の転写を制御することによりこの段階に作用していると仮定した.IRF8ノックアウトマウスに由来するマクロファージおよび樹状細胞におけるαVβ8インテグリン遺伝子のmRNAの発現を野生型マウスと比較したところ,野生型マウスでは発現が高かったのに対しIRF8ノックアウトマウスでは発現は非常に低かった.さらに,野生型マウスの骨髄に由来する樹状細胞およびマクロファージをインターフェロンγおよびリポ多糖により刺激するとαVβ8インテグリン遺伝子の転写が誘導されたのに対し,IRF8ノックアウトマウスではその誘導は軽微であった.αVβ8インテグリン遺伝子プロモーターの断片を含むレポーターベクターおよびIRF8の発現ベクターを293T細胞株に共導入して行ったルシフェラーゼアッセイにより,IRF8がαVβ8インテグリン遺伝子プロモーターの活性を増強させ,それにはプロモーター領域の-1081~-1952の領域が必要であることが示唆された.この領域にはインターフェロン応答配列に似た複数のIRF8結合予想部位が含まれており,そのひとつに変異を導入したところレポーター活性が失われたことから,αVβ8インテグリン遺伝子の転写におけるこの部位の関与が裏づけられた.さらに,内在性のIRF8遺伝子プロモーターによりIRF8-GFP融合タンパク質を発現するトランスジェニックマウスの骨髄に由来する樹状細胞に対し,GFPおよびIRF8に対する抗体を用いてクロマチン免疫沈降(chromatin immunoprecipitation:ChIP)法を行った結果,IRF8がルシフェラーゼアッセイにおいて推定された部位と結合することが確認された.以上から,IRF8がαVβ8インテグリン遺伝子の転写活性を直接に増強していることが明らかになった.
7.IRF8はTGFβを活性化することによりTh17細胞および制御性T細胞を分化させる
IRF8が抗原提示細胞によるTGFβシグナル伝達系の活性化を介してナイーブT細胞のエフェクターT細胞への分化に関与しているのかどうかを調べるため,CD11c陽性の樹状細胞とナイーブT細胞をin vitroにおいて生物学的な活性をもたない潜在型TGFβの存在のもとで共培養した.その結果,野生型の樹状細胞はTh17細胞への分化を促進したが,IRF8を欠損した樹状細胞は促進しなかった.一方,活性型TGFβの存在のもとでは野生型の樹状細胞およびIRF8を欠損した樹状細胞はともにTh17細胞へと分化したことから,IRF8を欠損した樹状細胞は潜在型TGFβを活性化することのできないことが示唆された.IRF8を欠損した樹状細胞の培養上清に含まれるインターロイキン17の量についても,潜在型TGFβの存在のもとでは低値であったが活性型TGFβの存在のもとでは野生型の樹状細胞と同等であった.さらに,IRF8を欠損した樹状細胞は潜在型TGFβの存在のもとでは制御性T細胞への分化を促進しなかったのに対し,野生型の樹状細胞はこれを促進したことから,IRF8を欠損した樹状細胞においてナイーブT細胞からエフェクター細胞へと分化できない原因はTGFβシグナル伝達系の欠損にあるという考えが裏づけられた.
TGFβシグナル伝達系により活性化されるSMADファミリー転写因子のうち,SMAD2はTh17細胞の活性化において重要とされている12).野生型の樹状細胞は潜在型TGFβの存在のもとでT細胞においてSMAD2のリン酸化を増強したのに対し,IRF8を欠損した樹状細胞は増強しなかった.その一方で,活性型TGFβの存在のもとでは野生型の樹状細胞およびIRF8を欠損した樹状細胞はともにSMAD2のリン酸化を増強した.in vivoにおいても,野生型マウスでは実験的自己免疫性脳脊髄炎の発症においてCD4陽性T細胞におけるSMAD2のリン酸化が増強したのに対し,IRF8ノックアウトマウスでは増強しなかった.さらに,IRF8およびαVβ8インテグリンをコードするmRNAの量が実験的自己免疫性脳脊髄炎を発症している野生型マウスの樹状細胞においては増加していたのに対し,IRF8ノックアウトマウスの樹状細胞では増加していなかった.実験的自己免疫性脳脊髄炎を発症している野生型マウスと比較して,IRF8ノックアウトマウスでは血清における活性型TGFβの濃度は非常に低値であった.これにくわえて,TGFβおよびインターロイキン6をコードするmRNAの量は実験的自己免疫性脳脊髄炎を発症している野生型マウスとIRF8ノックアウトマウスとは同等であったことから,IRF8ノックアウトマウスでは生物学的な活性をもつ活性型TGFβの産生が選択的に障害されていることが示された.
おわりに
この研究により,筆者らは,多発性硬化症のモデルである実験的自己免疫性脳脊髄炎を発症したマウスにおいて,抗原提示細胞におけるαVβ8インテグリンを介したTGFβの活性化からナイーブT細胞の感作によるエフェクター細胞への分化にいたる過程にて,転写因子IRF8が多面的に必要であるという強力で新しい証拠を得た(図1).IRF8遺伝子は多発性硬化症の疾患感受性遺伝子として同定されているが,その責任SNPは3’側非コード領域に位置する.したがって,感受性の原因としてはIRF8遺伝子の転写制御における異常が考えられるが,現在のところ,このSNPがIRF8遺伝子の転写にどのような影響をあたえているのかは不明である.さらに,多発性硬化症に対する現在の治療法がIRF8の発現や機能に対しどのような影響をあたえているのか,あるいは,Th17細胞が関与するとされるほかの自己免疫疾患においても同様の作用によりIRF8が病態に影響をあたえているのかなど,明らかにすべき点は多く残されており,この分野におけるこれからの研究が期待される.
文 献
- Acharya, M., Mukhopadhyay, S., Paidassi, H. et al.: αv Integrin expression by DCs is required for Th17 cell differentiation and development of experimental autoimmune encephalomyelitis in mice. J. Clin. Invest., 120, 4445-4452 (2010)[PubMed]
- Melton, A. C., Bailey-Bucktrout, S. L., Travis, M. A. et al.: Expression of αvβ8 integrin on dendritic cells regulates Th17 cell development and experimental autoimmune encephalomyelitis in mice. J. Clin. Invest., 120, 4436-4444 (2010)[PubMed]
- Ivanov, I. I., McKenzie, B. S., Zhou, L. et al.: The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell, 126, 1121-1133 (2006)[PubMed]
- Komiyama, Y., Nakae, S., Matsuki, T. et al.: IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J. Immunol., 177, 566-573 (2006)[PubMed]
- Axtell, R. C., de Jong, B. A., Boniface, K. et al.: T helper type 1 and 17 cells determine efficacy of interferon-β in multiple sclerosis and experimental encephalomyelitis. Nat. Med., 16, 406-412 (2010)[PubMed]
- De Jager, P. L., Jia, X., Wang, J. et al.: Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat. Genet., 41, 776-782 (2009)[PubMed]
- Tailor, P., Tamura, T., Morse, H. C. 3rd et al.: The BXH2 mutation in IRF8 differentially impairs dendritic cell subset development in the mouse. Blood, 111, 1942-1945 (2008)[PubMed]
- Hambleton, S., Salem, S., Bustamante, J. et al.: IRF8 mutations and human dendritic-cell immunodeficiency. N. Engl. J. Med., 365, 127-138 (2011)[PubMed]
- Bettelli, E., Carrier, Y., Gao, W. et al.: Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature, 441, 235-238 (2006)[PubMed]
- Park, H., Li, Z., Yang, X. O. et al.: A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol., 6, 1133-1141 (2005)[PubMed]
- Vignali, D. A. & Kuchroo, V. K.: IL-12 family cytokines: immunological playmakers. Nat. Immunol., 13, 722-728 (2012)[PubMed]
- Malhotra, N., Robertson, E. & Kang, J.: SMAD2 is essential for TGFβ-mediated Th17 cell generation. J. Biol. Chem., 285, 29044-29048 (2010)[PubMed]
著者プロフィール
略歴:2006年 横浜市立大学大学院医学研究科博士課程 修了,同年 横浜市立大学附属病院 常勤特別職,2007年 米国NIH National Institute of Child Health and Human Developmentポストドクトラルビジティングフェローを経て,2010年より横浜市立大学大学院医学研究科 特任助教(現 助教).
研究テーマ:全身性自己免疫疾患の発症の機序と臨床への応用.
尾里 啓子(Keiko Ozato)
米国NIH National Institute of Child Health and Human Development部門主任.
研究室URL:https://science.nichd.nih.gov/confluence/display/pgd/Keiko+Ozato+Lab
© 2014 吉見竜介・尾里啓子 Licensed under CC 表示 2.1 日本
