ミネラルコルチコイド受容体のリン酸化によりリガンドとの結合が制御され細胞の外液変化に対応した腎臓の応答が誘導される
柴田 茂
(米国Yale大学School of Medicine,Department of Genetics)
email:柴田 茂
DOI: 10.7875/first.author.2013.149
Mineralocorticoid receptor phosphorylation regulates ligand binding and renal response to volume depletion and hyperkalemia.
Shigeru Shibata, Jesse Rinehart, Junhui Zhang, Gilbert Moeckel, María Castañeda-Bueno, Amy L. Stiegler, Titus J. Boggon, Gerardo Gamba, Richard P. Lifton
Cell Metabolism, 18, 660-671 (2013)
核内受容体はリガンド依存性の転写因子であり,十分な量のリガンドが存在すれば受容体への結合およびシグナルの活性化が起こると考えられている.ミネラルコルチコイド受容体のリガンドであるアルドステロンは体液量の減少および高カリウム血症において分泌されるが,その結果として起こる腎臓の反応はそれぞれにおいて大きく異なる.筆者らは,ミネラルコルチコイド受容体のリガンド結合ドメインに存在するリン酸化部位を同定し,この部位のリン酸化によりリガンドと受容体との結合およびシグナルの活性化が抑制されることを見い出した.このリン酸化は腎臓の間在細胞においてのみ認められ,体液量の減少および高カリウム血症において逆の方向に制御された.体液量の減少はアンジオテンシンIIを介してミネラルコルチコイド受容体の脱リン酸化を促進し,間在細胞においてミネラルコルチコイド受容体の活性化をひき起こすことにより,アルドステロンの作用をK+の排泄からNaClの再吸収へとスイッチさせると考えられた.この研究から,翻訳後修飾によるリガンド結合能の可逆的な制御という核内受容体における新たな制御機構が明らかになり,体液量の減少および高カリウム血症それぞれに対する腎臓の適切な応答は,間在細胞におけるミネラルコルチコイド受容体のリン酸化の制御によりなされることが示された.
核内受容体は脂溶性の生理活性物質をリガンドとし,標的遺伝子の発現を制御することにより生命活動の維持に重要な役割を担っている.核内受容体のうちステロイド受容体は,リガンドと結合していない状態では熱ショックタンパク質のひとつであるHSP90などのシャペロンと結合して細胞質に存在する1).リガンドとの結合によりHSP90から解離したステロイド受容体は核へと移行し,標的とする特異的な塩基配列と結合して転写因子複合体を形成し標的遺伝子の転写を制御する2,3).リガンドと核内受容体との結合はシグナルの活性化の分子スイッチとみなすことができるが,これまでのモデルでは,十分な量のリガンドおよび受容体とが存在すれば,両者の結合およびシグナルの活性化がひき起こされると考えられてきた.
ミネラルコルチコイド受容体は腎臓や脳海馬,心血管系などに存在し,ステロイドホルモンであるアルドステロンとの結合により活性化する.アルドステロンは副腎の球状層において産生されており,おもに体液量の減少および高カリウム血症において分泌が刺激される.前者の場合はアンジオテンシンIIの作用により,後者の場合には血清におけるK+濃度の上昇にともなう球状層細胞の細胞膜の脱分極により,アルドステロンの産生が亢進する.
ミネラルコルチコイド受容体は腎臓のなかでも遠位ネフロンに高度に発現しており,アルドステロンの作用をうけて集合管の主細胞の上皮性Na+チャネルを介したNa+の再吸収を誘導する.この作用は起電性であり,体液量の減少したときにはNa+とともにCl-の流入(NaClの再吸収)が促進され,一方で,高カリウム血症ではNa+とひき換えにK+の排泄が誘導される.K+の排泄はおもにK+チャネルROMKの作用による.Cl-の輸送経路としては細胞間の経路や遠位尿細管細胞に存在するNa-Cl共輸送体のほか,間在細胞の重要性も新たに示されており,α型間在細胞のH+-ATPaseとβ型間在細胞のCl-/HCO3-交換輸送体であるPendrinが共役して作用することによりCl-を能動輸送している4,5).
それでは,アルドステロンの作用をうけた腎臓は,どのようにして体液量の減少ではK+の排泄を増加させずにNaClの再吸収を促進し,その逆に,高カリウム血症ではK+の排泄のみを最大化するのだろうか(図1)? 臨床においても,ミネラルコルチコイド受容体の阻害薬による降圧効果と副作用である血清K+濃度の上昇の程度とは相関に乏しく,アルドステロンの腎臓における作用を局所で制御するなんらかの機構の存在が示唆されていたが,その分子機構は不明であった.

筆者らは,これまで,ミネラルコルチコイド受容体の局所における制御機構の異常が高血圧や慢性腎臓病の発症にかかわることを明らかにしてきた6,7).この論文では,アルドステロンの生理的な作用をミネラルコルチコイド受容体の制御機構の観点から解析し,リガンド結合ドメインにおけるリン酸化によりリガンド結合能が可逆的に抑制されることを見い出した.
ヒトのミネラルコルチコイド受容体を腎臓に由来するCOS7細胞において発現し精製したのち,質量分析計を用いてリン酸化部位を解析し,その全長において16個のリン酸化部位(うち,14個が新規)を同定した.疑似リン酸化変異あるいは疑似非リン酸化変異の導入およびルシフェラーゼアッセイによるスクリーニングをへて,リガンド結合ドメインに存在するリン酸化部位Ser843に着目した.詳細な解析により,この部位のリン酸化されたミネラルコルチコイド受容体はリガンドとの結合が高度に障害され,その結果,生理的な濃度のリガンド存在のもとでも核への移行や転写の活性化が完全に抑制されることが明らかになった.
Ser843のリン酸化されたミネラルコルチコイド受容体の生体における意義を検討するため,この部位のリン酸化に特異的な抗体を作製し,特異性の確認ののち,マウスの組織において局在性を検討した.その結果,リン酸化をうけたミネラルコルチコイド受容体は腎臓にのみ存在が確認され,脳や心血管系には認められなかった.また驚くべきことに,腎臓では集合管の主細胞には認められず,隣接する間在細胞(α型とβ型の両者)の細胞質にのみ存在していた.
体液量の減少,高カリウム摂取,酸の負荷といった生理的な刺激に対するリン酸化の変化について検討したところ,体液量の減少によりその存在量は低下し,逆に,高カリウム血症においていちじるしく増加することが明らかになった.一方で,酸の負荷により変化はまったく認められなかった.これらのことから,K+の排泄が必要とされるときには間在細胞においてミネラルコルチコイド受容体は抑制され,一方で,体液量の減少したときにはSer843が脱リン酸化をうけることによりその活性は回復すると考えられた.
体液量の減少はレニン-アンジオテンシン系を活性化しアンジオテンシンIIの産生を促進する.この作用は高カリウム血症においては認められず,アンジオテンシンIIは両者を区別する重要なシグナルと考えられた.アンジオテンシンII受容体のうちAT1受容体は間在細胞にも存在していることから,ミネラルコルチコイド受容体の脱リン酸化におけるアンジオテンシンIIのシグナルの役割について検討した.AT1受容体を欠損したマウスと野生型のマウスとで体液量の減少に対するリン酸化の変化について比較したところ,野生型マウスで認められた脱リン酸化はAT1受容体を欠損したマウスでは完全に消失していた.また,野生型マウスに対しアンジオテンシンIIを投与したところリン酸化をうけたミネラルコルチコイド受容体は顕著に減少したことから,体液量の減少においてはアンジオテンシンIIのシグナルによりミネラルコルチコイド受容体のSer843における脱リン酸化がひき起こされることが明らかになった.
つぎに,下流のタンパク質について解析した.その生理的な役割から,ミネラルコルチコイド受容体のターゲットは間在細胞に存在するイオン輸送体である可能性が高いと思われた.そこで,間在細胞に特異的に存在する膜輸送体に焦点をあて解析したところ,H+-ATPaseのB1サブユニットおよびCl-/HCO3-交換輸送体であるPendrinの発現がミネラルコルチコイド受容体の脱リン酸化にともない上昇し,逆に,リン酸化をうけたミネラルコルチコイド受容体の増加とともに低下することがわかった.さらに,これらの膜輸送体の発現の上昇はミネラルコルチコイド受容体の阻害薬により抑制されたことから,H+-ATPaseのB1サブユニットおよびPendrinがSer843のリン酸化とミネラルコルチコイド受容体の標的タンパク質であることが示された.間在細胞の頂端膜に存在するH+-ATPaseおよびPendrinはCl-の再吸収にかかわることが明らかにされており4,5),Ser843のリン酸化は間在細胞に選択的にミネラルコルチコイド受容体の活性を制御することによりH+-ATPaseのB1サブユニットおよびPendrinの発現を制御し,Cl-の再吸収を制御していると考えられた(図2a).
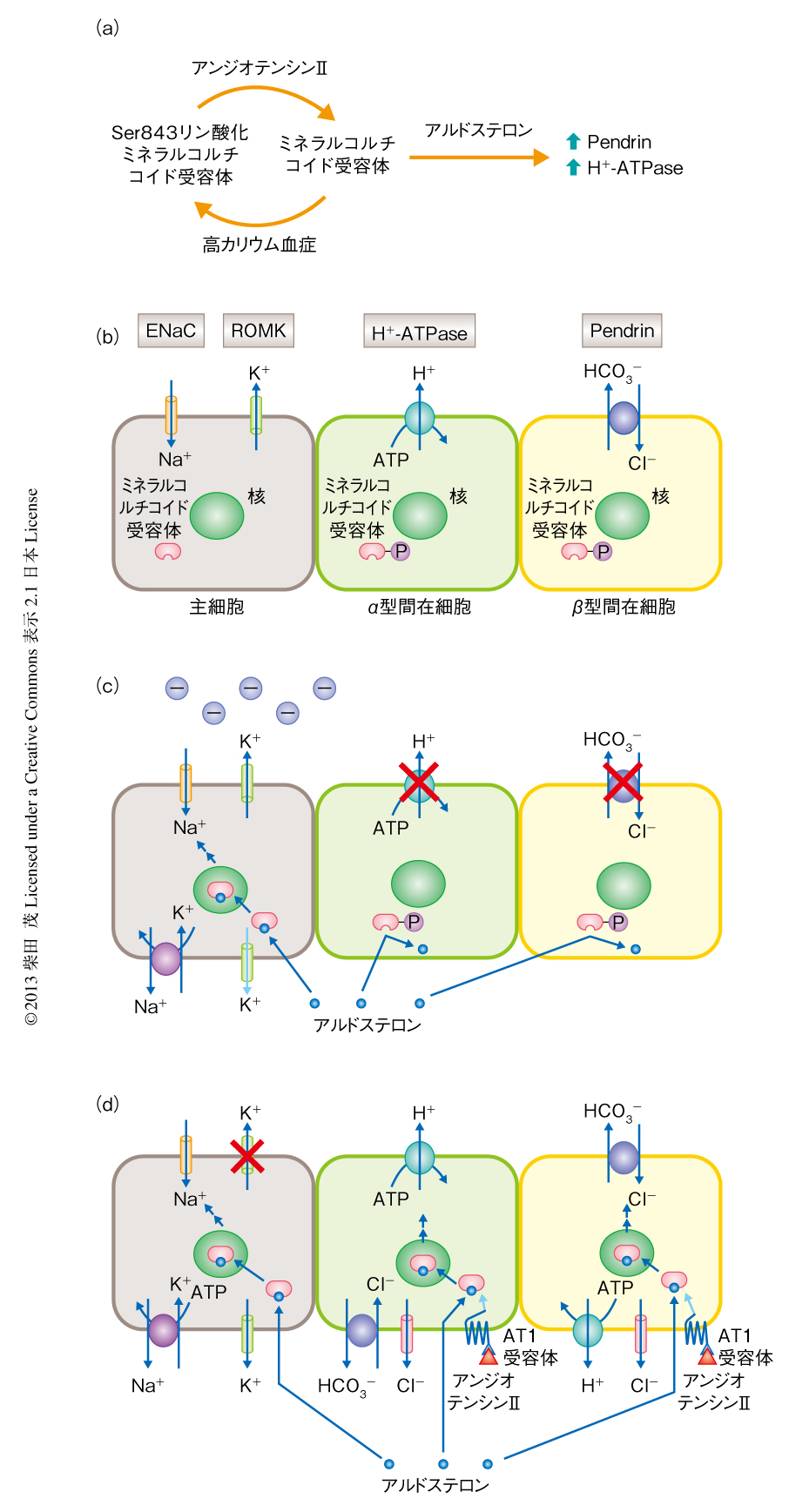
単遺伝子の異常による高血圧の多くは低カリウム血症を呈するが,偽性低アルドステロン症II型では例外的に高カリウム血症をともなう.この疾患の原因としてWNKキナーゼの遺伝子変異が報告されており8),偽性低アルドステロン症II型はWNKキナーゼの機能の異常により腎臓におけるNaClの再吸収とK+の排泄とのバランスが障害される(すなわち,K+の排泄が抑制されNaClの再吸収が亢進する)ことがその本態と考えられている9).これまでの結果から,ミネラルコルチコイド受容体のSer843におけるリン酸化によりアルドステロンのNaCl再吸収作用とK+作用とのバランスが制御される(図2 b-d)と考えられたことから,偽性低アルドステロン症II型のマウスモデルにおけるSer843のリン酸化の量について検討した.WNKキナーゼのアイソザイムであるWNK4は間在細胞においても発現が確認され,興味深いことに,偽性低アルドステロン症II型モデルマウスではリン酸化をうけたミネラルコルチコイド受容体の顕著な低下が認められた.そこで,このモデルマウスに対しアルドステロンを投与し,間在細胞におけるミネラルコルチコイド受容体の反応を検討したところ,野生型のマウスと比較し偽性低アルドステロン症II型モデルマウスではH+-ATPaseのB1サブユニットおよびPendrinの発現が高度に誘導された.このことから,ミネラルコルチコイド受容体の脱リン酸化にWNK4が関与することが確認され,偽性低アルドステロン症II型の病態にミネラルコルチコイド受容体のSer843におけるリン酸化の異常が関与することが示唆された.また,WNK4の下流においてPP1ホスファターゼの作用することが報告されているが10),in vitroにおける脱リン酸化解析により,PP1ホスファターゼがSer843のリン酸化されたミネラルコルチコイド受容体の脱リン酸化酵素として作用することが明らかになった.
この論文においては,リガンド結合ドメインにおけるリン酸化によりミネラルコルチコイド受容体のリガンドとの結合能およびシグナルの活性化が細胞に選択的に制御されることを報告した.この部位の脱リン酸化により間在細胞を介するCl-の再吸収の経路が活性化され,ミネラルコルチコイド受容体の腎臓における作用がK+の排泄からNaClの再吸収へとスイッチすると考えられた(図2).ミネラルコルチコイド受容体においてリガンド結合ドメインの翻訳後修飾によりリガンドとの結合能が可逆的に制御されるというモデルは,核内受容体の制御機構の新たな一面を明らかにするものであり,類似した分子機構がほかの組織のミネラルコルチコイド受容体,あるいは,ほかの核内受容体における活性制御にかかわる可能性がある.
略歴:2007年 東京大学大学院医学系研究科 修了,同 博士研究員,米国Yale大学School of Medicine研究員を経て,2013年より同Associate Research Scientist.
研究テーマ:慢性腎臓病の進展の機構,翻訳後修飾による腎臓の機能の制御.
© 2013 柴田 茂 Licensed under CC 表示 2.1 日本
(米国Yale大学School of Medicine,Department of Genetics)
email:柴田 茂
DOI: 10.7875/first.author.2013.149
Mineralocorticoid receptor phosphorylation regulates ligand binding and renal response to volume depletion and hyperkalemia.
Shigeru Shibata, Jesse Rinehart, Junhui Zhang, Gilbert Moeckel, María Castañeda-Bueno, Amy L. Stiegler, Titus J. Boggon, Gerardo Gamba, Richard P. Lifton
Cell Metabolism, 18, 660-671 (2013)
この論文に出現する遺伝子・タンパク質のUniprot ID
ミネラルコルチコイド受容体(P08235), Mineralocorticoid receptor(P08235), 核内受容体, ステロイド受容体, HSP90, ROMK(P48048), H+-ATPase, Pendrin(O43511), AT1受容体(P29754), WNKキナーゼ, PP1ホスファターゼ, WNK4(Q80UE6), ENaC
要 約
核内受容体はリガンド依存性の転写因子であり,十分な量のリガンドが存在すれば受容体への結合およびシグナルの活性化が起こると考えられている.ミネラルコルチコイド受容体のリガンドであるアルドステロンは体液量の減少および高カリウム血症において分泌されるが,その結果として起こる腎臓の反応はそれぞれにおいて大きく異なる.筆者らは,ミネラルコルチコイド受容体のリガンド結合ドメインに存在するリン酸化部位を同定し,この部位のリン酸化によりリガンドと受容体との結合およびシグナルの活性化が抑制されることを見い出した.このリン酸化は腎臓の間在細胞においてのみ認められ,体液量の減少および高カリウム血症において逆の方向に制御された.体液量の減少はアンジオテンシンIIを介してミネラルコルチコイド受容体の脱リン酸化を促進し,間在細胞においてミネラルコルチコイド受容体の活性化をひき起こすことにより,アルドステロンの作用をK+の排泄からNaClの再吸収へとスイッチさせると考えられた.この研究から,翻訳後修飾によるリガンド結合能の可逆的な制御という核内受容体における新たな制御機構が明らかになり,体液量の減少および高カリウム血症それぞれに対する腎臓の適切な応答は,間在細胞におけるミネラルコルチコイド受容体のリン酸化の制御によりなされることが示された.
はじめに
核内受容体は脂溶性の生理活性物質をリガンドとし,標的遺伝子の発現を制御することにより生命活動の維持に重要な役割を担っている.核内受容体のうちステロイド受容体は,リガンドと結合していない状態では熱ショックタンパク質のひとつであるHSP90などのシャペロンと結合して細胞質に存在する1).リガンドとの結合によりHSP90から解離したステロイド受容体は核へと移行し,標的とする特異的な塩基配列と結合して転写因子複合体を形成し標的遺伝子の転写を制御する2,3).リガンドと核内受容体との結合はシグナルの活性化の分子スイッチとみなすことができるが,これまでのモデルでは,十分な量のリガンドおよび受容体とが存在すれば,両者の結合およびシグナルの活性化がひき起こされると考えられてきた.
ミネラルコルチコイド受容体は腎臓や脳海馬,心血管系などに存在し,ステロイドホルモンであるアルドステロンとの結合により活性化する.アルドステロンは副腎の球状層において産生されており,おもに体液量の減少および高カリウム血症において分泌が刺激される.前者の場合はアンジオテンシンIIの作用により,後者の場合には血清におけるK+濃度の上昇にともなう球状層細胞の細胞膜の脱分極により,アルドステロンの産生が亢進する.
ミネラルコルチコイド受容体は腎臓のなかでも遠位ネフロンに高度に発現しており,アルドステロンの作用をうけて集合管の主細胞の上皮性Na+チャネルを介したNa+の再吸収を誘導する.この作用は起電性であり,体液量の減少したときにはNa+とともにCl-の流入(NaClの再吸収)が促進され,一方で,高カリウム血症ではNa+とひき換えにK+の排泄が誘導される.K+の排泄はおもにK+チャネルROMKの作用による.Cl-の輸送経路としては細胞間の経路や遠位尿細管細胞に存在するNa-Cl共輸送体のほか,間在細胞の重要性も新たに示されており,α型間在細胞のH+-ATPaseとβ型間在細胞のCl-/HCO3-交換輸送体であるPendrinが共役して作用することによりCl-を能動輸送している4,5).
それでは,アルドステロンの作用をうけた腎臓は,どのようにして体液量の減少ではK+の排泄を増加させずにNaClの再吸収を促進し,その逆に,高カリウム血症ではK+の排泄のみを最大化するのだろうか(図1)? 臨床においても,ミネラルコルチコイド受容体の阻害薬による降圧効果と副作用である血清K+濃度の上昇の程度とは相関に乏しく,アルドステロンの腎臓における作用を局所で制御するなんらかの機構の存在が示唆されていたが,その分子機構は不明であった.
筆者らは,これまで,ミネラルコルチコイド受容体の局所における制御機構の異常が高血圧や慢性腎臓病の発症にかかわることを明らかにしてきた6,7).この論文では,アルドステロンの生理的な作用をミネラルコルチコイド受容体の制御機構の観点から解析し,リガンド結合ドメインにおけるリン酸化によりリガンド結合能が可逆的に抑制されることを見い出した.
1.リガンド結合ドメインにおけるリン酸化はミネラルコルチコイド受容体の活性化を抑制する
ヒトのミネラルコルチコイド受容体を腎臓に由来するCOS7細胞において発現し精製したのち,質量分析計を用いてリン酸化部位を解析し,その全長において16個のリン酸化部位(うち,14個が新規)を同定した.疑似リン酸化変異あるいは疑似非リン酸化変異の導入およびルシフェラーゼアッセイによるスクリーニングをへて,リガンド結合ドメインに存在するリン酸化部位Ser843に着目した.詳細な解析により,この部位のリン酸化されたミネラルコルチコイド受容体はリガンドとの結合が高度に障害され,その結果,生理的な濃度のリガンド存在のもとでも核への移行や転写の活性化が完全に抑制されることが明らかになった.
2.ミネラルコルチコイド受容体のSer843は高カリウム血症ではリン酸化され逆に体液量の減少では脱リン酸化される
Ser843のリン酸化されたミネラルコルチコイド受容体の生体における意義を検討するため,この部位のリン酸化に特異的な抗体を作製し,特異性の確認ののち,マウスの組織において局在性を検討した.その結果,リン酸化をうけたミネラルコルチコイド受容体は腎臓にのみ存在が確認され,脳や心血管系には認められなかった.また驚くべきことに,腎臓では集合管の主細胞には認められず,隣接する間在細胞(α型とβ型の両者)の細胞質にのみ存在していた.
体液量の減少,高カリウム摂取,酸の負荷といった生理的な刺激に対するリン酸化の変化について検討したところ,体液量の減少によりその存在量は低下し,逆に,高カリウム血症においていちじるしく増加することが明らかになった.一方で,酸の負荷により変化はまったく認められなかった.これらのことから,K+の排泄が必要とされるときには間在細胞においてミネラルコルチコイド受容体は抑制され,一方で,体液量の減少したときにはSer843が脱リン酸化をうけることによりその活性は回復すると考えられた.
3.アンジオテンシンIIのシグナルはミネラルコルチコイド受容体を脱リン酸化する
体液量の減少はレニン-アンジオテンシン系を活性化しアンジオテンシンIIの産生を促進する.この作用は高カリウム血症においては認められず,アンジオテンシンIIは両者を区別する重要なシグナルと考えられた.アンジオテンシンII受容体のうちAT1受容体は間在細胞にも存在していることから,ミネラルコルチコイド受容体の脱リン酸化におけるアンジオテンシンIIのシグナルの役割について検討した.AT1受容体を欠損したマウスと野生型のマウスとで体液量の減少に対するリン酸化の変化について比較したところ,野生型マウスで認められた脱リン酸化はAT1受容体を欠損したマウスでは完全に消失していた.また,野生型マウスに対しアンジオテンシンIIを投与したところリン酸化をうけたミネラルコルチコイド受容体は顕著に減少したことから,体液量の減少においてはアンジオテンシンIIのシグナルによりミネラルコルチコイド受容体のSer843における脱リン酸化がひき起こされることが明らかになった.
4.ミネラルコルチコイド受容体のSer843におけるリン酸化により間在細胞によるCl-の再吸収が制御される
つぎに,下流のタンパク質について解析した.その生理的な役割から,ミネラルコルチコイド受容体のターゲットは間在細胞に存在するイオン輸送体である可能性が高いと思われた.そこで,間在細胞に特異的に存在する膜輸送体に焦点をあて解析したところ,H+-ATPaseのB1サブユニットおよびCl-/HCO3-交換輸送体であるPendrinの発現がミネラルコルチコイド受容体の脱リン酸化にともない上昇し,逆に,リン酸化をうけたミネラルコルチコイド受容体の増加とともに低下することがわかった.さらに,これらの膜輸送体の発現の上昇はミネラルコルチコイド受容体の阻害薬により抑制されたことから,H+-ATPaseのB1サブユニットおよびPendrinがSer843のリン酸化とミネラルコルチコイド受容体の標的タンパク質であることが示された.間在細胞の頂端膜に存在するH+-ATPaseおよびPendrinはCl-の再吸収にかかわることが明らかにされており4,5),Ser843のリン酸化は間在細胞に選択的にミネラルコルチコイド受容体の活性を制御することによりH+-ATPaseのB1サブユニットおよびPendrinの発現を制御し,Cl-の再吸収を制御していると考えられた(図2a).
5.ミネラルコルチコイド受容体の脱リン酸化にはWNKキナーゼとPP1ホスファターゼが関与する
単遺伝子の異常による高血圧の多くは低カリウム血症を呈するが,偽性低アルドステロン症II型では例外的に高カリウム血症をともなう.この疾患の原因としてWNKキナーゼの遺伝子変異が報告されており8),偽性低アルドステロン症II型はWNKキナーゼの機能の異常により腎臓におけるNaClの再吸収とK+の排泄とのバランスが障害される(すなわち,K+の排泄が抑制されNaClの再吸収が亢進する)ことがその本態と考えられている9).これまでの結果から,ミネラルコルチコイド受容体のSer843におけるリン酸化によりアルドステロンのNaCl再吸収作用とK+作用とのバランスが制御される(図2 b-d)と考えられたことから,偽性低アルドステロン症II型のマウスモデルにおけるSer843のリン酸化の量について検討した.WNKキナーゼのアイソザイムであるWNK4は間在細胞においても発現が確認され,興味深いことに,偽性低アルドステロン症II型モデルマウスではリン酸化をうけたミネラルコルチコイド受容体の顕著な低下が認められた.そこで,このモデルマウスに対しアルドステロンを投与し,間在細胞におけるミネラルコルチコイド受容体の反応を検討したところ,野生型のマウスと比較し偽性低アルドステロン症II型モデルマウスではH+-ATPaseのB1サブユニットおよびPendrinの発現が高度に誘導された.このことから,ミネラルコルチコイド受容体の脱リン酸化にWNK4が関与することが確認され,偽性低アルドステロン症II型の病態にミネラルコルチコイド受容体のSer843におけるリン酸化の異常が関与することが示唆された.また,WNK4の下流においてPP1ホスファターゼの作用することが報告されているが10),in vitroにおける脱リン酸化解析により,PP1ホスファターゼがSer843のリン酸化されたミネラルコルチコイド受容体の脱リン酸化酵素として作用することが明らかになった.
おわりに
この論文においては,リガンド結合ドメインにおけるリン酸化によりミネラルコルチコイド受容体のリガンドとの結合能およびシグナルの活性化が細胞に選択的に制御されることを報告した.この部位の脱リン酸化により間在細胞を介するCl-の再吸収の経路が活性化され,ミネラルコルチコイド受容体の腎臓における作用がK+の排泄からNaClの再吸収へとスイッチすると考えられた(図2).ミネラルコルチコイド受容体においてリガンド結合ドメインの翻訳後修飾によりリガンドとの結合能が可逆的に制御されるというモデルは,核内受容体の制御機構の新たな一面を明らかにするものであり,類似した分子機構がほかの組織のミネラルコルチコイド受容体,あるいは,ほかの核内受容体における活性制御にかかわる可能性がある.
文 献
- Pratt, W. B.: The role of the hsp90-based chaperone system in signal transduction by nuclear receptors and receptors signaling via MAP kinase. Annu. Rev. Pharmacol. Toxicol., 37, 297-326 (1997)[PubMed]
- Mangelsdorf, D. J., Thummel, C., Beato, M. et al.: The nuclear receptor superfamily: the second decade. Cell, 83, 835-839 (1995)[PubMed]
- McKenna, N. J. & O'Malley, B. W.: Combinatorial control of gene expression by nuclear receptors and coregulators. Cell, 108, 465-474 (2002)[PubMed]
- Soleimani, M., Barone, S., Xu, J. et al.: Double knockout of pendrin and Na-Cl cotransporter (NCC) causes severe salt wasting, volume depletion, and renal failure. Proc. Natl. Acad. Sci. USA, 109, 13368-13373 (2012)[PubMed]
- Wall, S. M. & Pech, V.: The interaction of pendrin and the epithelial sodium channel in blood pressure regulation. Curr. Opin. Nephrol. Hypertens., 17, 18-24 (2008)[PubMed]
- Shibata, S., Nagase, M., Yoshida, S. et al.: Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat. Med., 14, 1370-1376 (2008)[PubMed]
- Shibata, S., Mu, S., Kawarazaki, H. et al.: Rac1 GTPase in rodent kidneys is essential for salt-sensitive hypertension via a mineralocorticoid receptor-dependent pathway. J. Clin. Invest., 121, 3233-3243 (2011)[PubMed]
- Wilson, F. H., Disse-Nicodeme, S., Choate, K. A. et al.: Human hypertension caused by mutations in WNK kinases. Science, 293, 1107-1112 (2001)[PubMed]
- Kahle, K. T., Ring, A. M., Lifton, R. P.: Molecular physiology of the WNK kinases. Annu. Rev. Physiol., 70, 329-355 (2008)[PubMed]
- Lin, D. H., Yue, P., Rinehart, J. et al.: Protein phosphatase 1 modulates the inhibitory effect of With-no-Lysine kinase 4 on ROMK channels. Am. J. Physiol. Renal Physiol., 303, F110-F119 (2012)[PubMed]
著者プロフィール
略歴:2007年 東京大学大学院医学系研究科 修了,同 博士研究員,米国Yale大学School of Medicine研究員を経て,2013年より同Associate Research Scientist.
研究テーマ:慢性腎臓病の進展の機構,翻訳後修飾による腎臓の機能の制御.
© 2013 柴田 茂 Licensed under CC 表示 2.1 日本
