ケモカイン受容体CXCR2を発現する骨髄由来抑制細胞の遊走および局所への浸潤は大腸炎に関連する腫瘍の形成において重要な役割を担う
加藤 弘・Dingzhi Wang・Raymond N. DuBois
(米国Arizona州立大学Biodesign Institute,Laboratory for Inflammation and Cancer)
email:加藤 弘
DOI: 10.7875/first.author.2013.147
CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis.
Hiroshi Katoh, Dingzhi Wang, Takiko Daikoku, Haiyan Sun, Sudhansu K. Dey, Raymond N. DuBois
Cancer Cell, 24, 631-644 (2013)
炎症性腸疾患などの慢性炎症は発がんおよび腫瘍の進展におけるリスク因子のひとつである.しかしながら,炎症および炎症性メディエーターの発がんにおける詳細な分子機構はいまだ解明されていない.この研究では,ケモカイン受容体であるCXCR2をノックアウトしたマウスを用いた大腸炎に関連する腫瘍のモデルマウスを用いて,CXCR2の発現が骨髄由来抑制細胞の炎症の局所への遊走に不可欠であることを明らかにした.CXCR2のリガンドとなるケモカインの産生は炎症を生じた大腸の粘膜および腫瘍において亢進しており,骨髄由来抑制細胞の局所への浸潤を促進していた.野生型の骨髄由来抑制細胞をCXCR2ノックアウトマウスへ移入することにより,炎症に関連する腫瘍の形成および進展は回復することが示された.また,局所に浸潤した骨髄由来抑制細胞はCD8陽性T細胞の細胞傷害活性を抑制することにより,腫瘍細胞の免疫回避を誘導していた.これらの発見が,炎症に関連する大腸腫瘍の進展の分子機構の解明にくわえ,腫瘍において免疫寛容を標的とする新規の診断法あるいは治療法につながることが期待される.
大腸がんは悪性腫瘍のうち世界第2位の罹患率,第4位の死亡数をもたらしているが,大腸内視鏡検査の普及にもかかわらず,いまだ半数以上の症例は初診のとき進行がんとしてみつかっている1).遺伝子の変異あるいはエピジェネティックな変異にくわえて,慢性炎症は大腸がんのリスク因子のひとつである2).実際に,潰瘍性大腸炎の20%以上の症例は診断ののち30年以内に大腸がんを発生することが報告されている3).このように慢性炎症と大腸がんには密接な関連がみられることから,大腸炎に関連する腫瘍の研究は“proof of concept”モデルとして,慢性炎症および炎症性メディエーターの腫瘍の発生,進展,転移における分子機構の解明につながると考えられる.
慢性炎症は損傷や病原微生物への暴露によりもたらされる免疫反応の亢進状態が持続することにより生じる.実際に,腸内細菌叢の重要性は炎症性腸疾患の患者において証明されている4).慢性大腸炎が腸内細菌叢の存在に依存していることは動物実験により直接的に示唆され5),インターロイキン10ノックアウトマウスを使用した炎症関連大腸がんモデルマウスは無菌状態では大腸がんを発生しない6).同様に,遺伝性および孤発性の大腸がんモデルマウスでも腸内細菌叢が腫瘍の増殖にかかわることが示唆されている7).注目すべきことに,病原細菌は炎症を起こした大腸の粘膜においてシクロオキシゲナーゼ2の発現を誘導し8),シクロオキシゲナーゼ2とそれに由来する炎症性メディエーターであるプロスタグランジンE2は炎症性腸疾患の症例の大腸の粘膜においていちじるしく増加している9).
炎症性腸疾患において,炎症性細胞の著明な浸潤は主要な組織学的な特徴のひとつである.同様に,炎症に関連する大腸がんおよび孤発性の大腸がんにおいてもさまざまな免疫細胞や内皮細胞が動員され,腫瘍微小環境を構築する10).ケモカインは循環血から炎症の局所あるいは腫瘍に炎症性細胞を動員するため不可欠である.腸の粘膜における炎症性ケモカインの増加と炎症性細胞の著明な浸潤は炎症性腸疾患において疾患の程度と密接に相関する.炎症性ケモカインは孤発性大腸がんの大腸上皮においてもいちじるしく亢進している11).しかしながら,こうしたケモカインおよびケモカイン受容体の炎症性腸疾患および大腸がんにおける詳細な分子機構はいまだ明らかになっていない.
がんの発生と進展は宿主の免疫監視からの腫瘍細胞の逃避にも依存している.大腸がんなど固形がんにおいては,Th1細胞とTh2細胞との不均衡や,骨髄由来抑制細胞(myeloid-derived suppressor cell:MDSC)あるいは制御性T細胞など免疫抑制細胞の亢進が生じている.血中の骨髄由来抑制細胞の数が大腸がんの臨床病期や遠隔転移と相関することが報告されている12).骨髄由来抑制細胞はT細胞やナチュラルキラー細胞の機能を抑制することによりがん細胞の免疫逃避に貢献する13).しかしながら,骨髄由来抑制細胞の循環血から局所への動員の分子機構や大腸がんの進展にかかわる詳細な機序についてはいまだ不明であった.
筆者らの研究室では,炎症性メディエーターであるプロスタグランジンE2が大腸がん細胞からのケモカインCXCL1の産生を直接に促進し,それにより腫瘍における血管の新生が誘導されることを報告した14).CXCL1はケモカイン受容体CXCR2のリガンドのひとつであり(図1a),CXCR2は好中球に発現しその炎症性の動員において中心的な役割を担うことが知られている15).炎症性腸疾患において血清のCXCL1の量は増加しており,疾患の程度と相関する16).同様に,CXCL1およびCXCR2の発現が孤発性大腸がんの組織において上昇していることを報告した14).この研究では,炎症に関連する大腸腫瘍におけるCXCR2シグナル伝達系の分子機構について検討した.
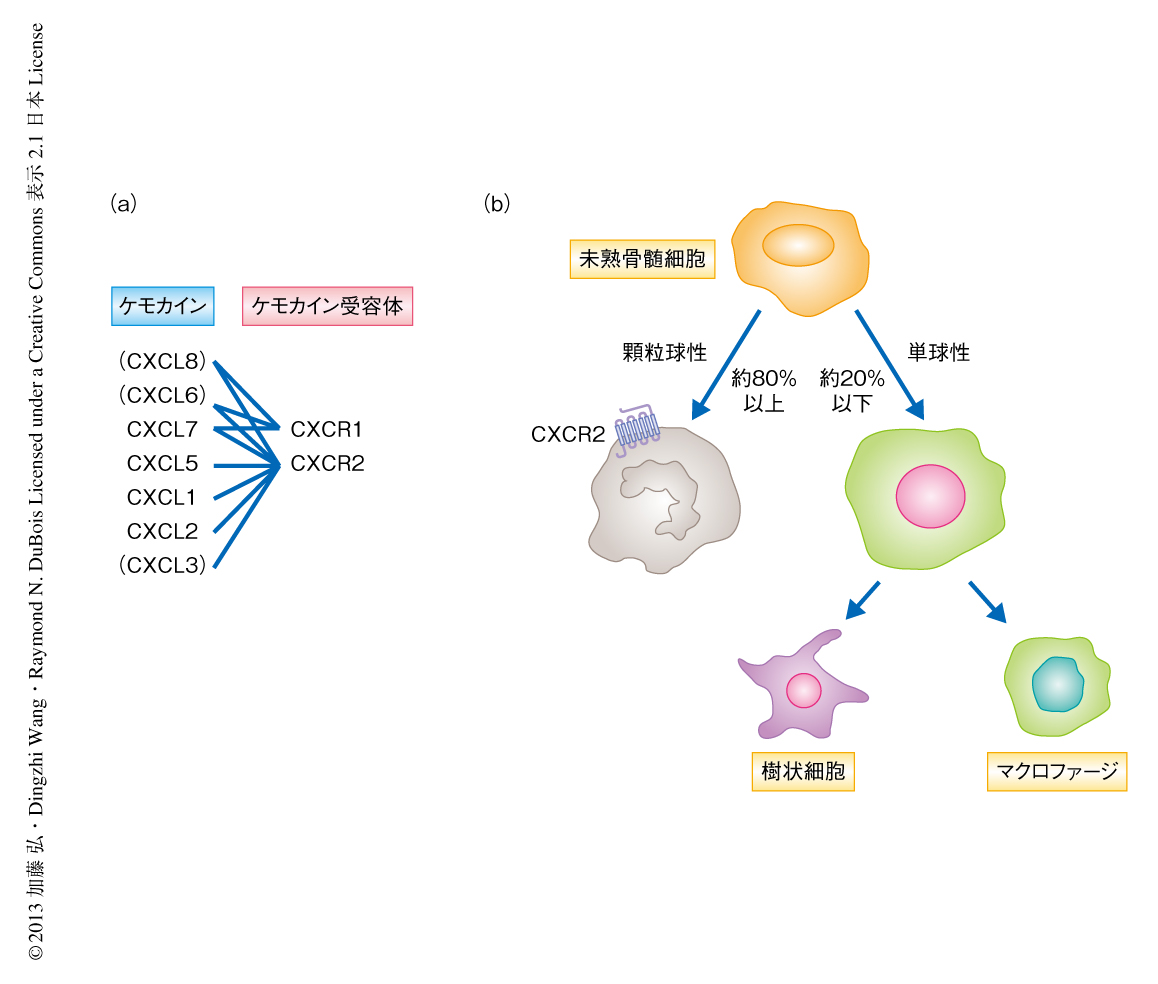
炎症に関連する大腸腫瘍の発症におけるCXCR2シグナル伝達系の役割を調べるため,野生型マウスおよびCXCR2ノックアウトマウスに大腸に特異的な発がん物質であるアゾキシメタンを腹腔内投与し,ひきつづき,大腸炎をひき起こす薬剤であるデキストラン硫酸ナトリウムを4サイクルにわたり服用させる,慢性大腸炎および炎症関連大腸腫瘍モデルマウスを使用した.この2つの薬剤の投与により,野生型マウスにおいては血便および大腸の短縮などの肉眼所見がみられ,組織的な炎症スコアの有意な上昇が認められた.CXCR2ノックアウトマウスではこれらの肉眼所見および組織的な炎症の発生は有意に抑制されていた.さらに,CXCR2ノックアウトマウスでは腫瘍細胞の増殖の抑制による炎症関連大腸腫瘍の劇的な減少が認められた.炎症の程度と腫瘍の数は明らかに相関していた.また,CXCR2ノックアウトマウスでは腫瘍における腺がんの比率も低下しており,CXCR2は腫瘍の進展を促進すると考えられた.以上から,ケモカイン受容体CXCR2は慢性大腸炎および炎症関連腫瘍の形成および進展に重要な役割をもつことが示唆された.
CXCR2が免疫細胞の動員に関与しているかどうかを調べるため,まず,デキストラン硫酸ナトリウムにより誘導した大腸炎モデルマウスにおける大腸炎症の組織において,免疫細胞のプロファイルを解析した.デキストラン硫酸ナトリウムの単回投与による急性炎症期ではさまざまな免疫細胞が局所に動員され,CXCR2の欠損により好中球,マクロファージ,骨髄由来抑制細胞の動員が抑制された.一方,デキストラン硫酸ナトリウムのくり返し投与による慢性炎症期ではCXCR2の欠損により骨髄由来抑制細胞の動員のみが有意に抑制された.
マウスでは,未熟な骨髄に由来する細胞(しばしば,骨髄由来抑制細胞)はCD11b陽性Gr-1陽性細胞として定義され,さらに,顆粒球性の骨髄由来抑制細胞(CD11b陽性Ly6G陽性細胞)と単球性の骨髄由来抑制細胞(CD11b陽性Ly6C陽性細胞)とに分けられる17)(図1b).大腸炎の組織では90%以上の骨髄由来抑制細胞が顆粒球性であった.興味深いことに,CXCR2ノックアウトマウスでは正常な状態および大腸炎の両方において循環血に顆粒球性の骨髄由来抑制細胞の蓄積が認められ,これは慢性炎症によりさらに蓄積した.このことから,CXCR2を欠損した骨髄由来抑制細胞は局所への浸潤能が欠損しているものと考えられた.注目すべきことに,血中の顆粒球性の骨髄由来抑制細胞の大部分はCXCR2を発現していたが,単球性の骨髄由来抑制細胞にはCXCR2の発現は認められなかった.慢性大腸炎モデルマウスと同様に,アゾキシメタンおよびデキストラン硫酸ナトリウムにより誘導した炎症関連大腸腫瘍においても顆粒球性の骨髄由来抑制細胞の大腸の組織への浸潤の亢進が認められ,腫瘍組織では周囲の炎症組織よりもさらに高度な浸潤がみられた.CXCR2の欠損により顆粒球性の骨髄由来抑制細胞の腫瘍組織および炎症組織の両方への動員は著明に抑制され,慢性炎症と同様の免疫細胞のプロファイルを示した.これを反映し,CXCR2ノックアウトマウスの血中には顆粒球性の骨髄由来抑制細胞の蓄積が認められた.一方,CXCR2ノックアウトマウスの循環血における顆粒球性の骨髄由来抑制細胞の生存率は野生型マウスより有意に低下していた.以上から,ケモカイン受容体CXCR2は顆粒球性の骨髄由来抑制細胞の循環血より炎症組織および腫瘍への動員に不可欠であることが示された.
以上の結果から,CXCR2を発現する免疫細胞の動員に不可欠であるCXCR2のリガンドとなるケモカインの発現は炎症組織および腫瘍組織において上昇していると予想された.実際に,野生型マウスでは大腸炎組織においてケモカインCXCL1,CXCL2,CXCL5の発現が上昇しており,腫瘍組織ではさらにこれらの発現が亢進していた.組織におけるCXCR2のリガンドとなるケモカインの発現と顆粒球性の骨髄由来抑制細胞の浸潤の程度とは相関していた.CXCR2のリガンドとなるケモカインを産生する細胞を同定するためin situハイブリダイゼーションを行ったところ,これらのmRNAはおもに大腸上皮,とくに,腫瘍細胞において発現していた.
CXCR2のリガンドとなるケモカインによる細胞遊走アッセイを行ったところ,野生型の顆粒球性の骨髄由来抑制細胞は遊走を示したがCXCR2を欠損した顆粒球性の骨髄由来抑制細胞には遊走が認められなかったことから,CXCR2がリガンドとなるケモカインに誘導される化学遊走に必須であることが証明された.
骨髄に由来する細胞はインターロイキン6の主要な産生細胞であり,インターロイキン6-Stat3シグナル伝達系は骨髄由来抑制細胞の免疫抑制機能において中心的な役割をはたす13).用いたモデルマウスにおいてインターロイキン6の発現は大腸炎を生じた粘膜および腫瘍組織において上昇しており,CXCR2ノックアウトマウスでは抑制されていた.血清におけるインターロイキン6の発現はCXCR2ノックアウトマウスにおいて亢進しており,骨髄由来抑制細胞と同様のパターンを示した.また,リン酸化Stat3の発現は野生型マウスの腫瘍間質細胞には認められたが,CXCR2ノックアウトマウスには認められなかった.したがって,大腸組織に浸潤した骨髄由来抑制細胞は免疫抑制作用をもつ可能性が示唆された.以上から,CXCR2リガンド-CXCR2シグナル伝達系は炎症組織および腫瘍組織への動員において必須であることが証明された(図2).
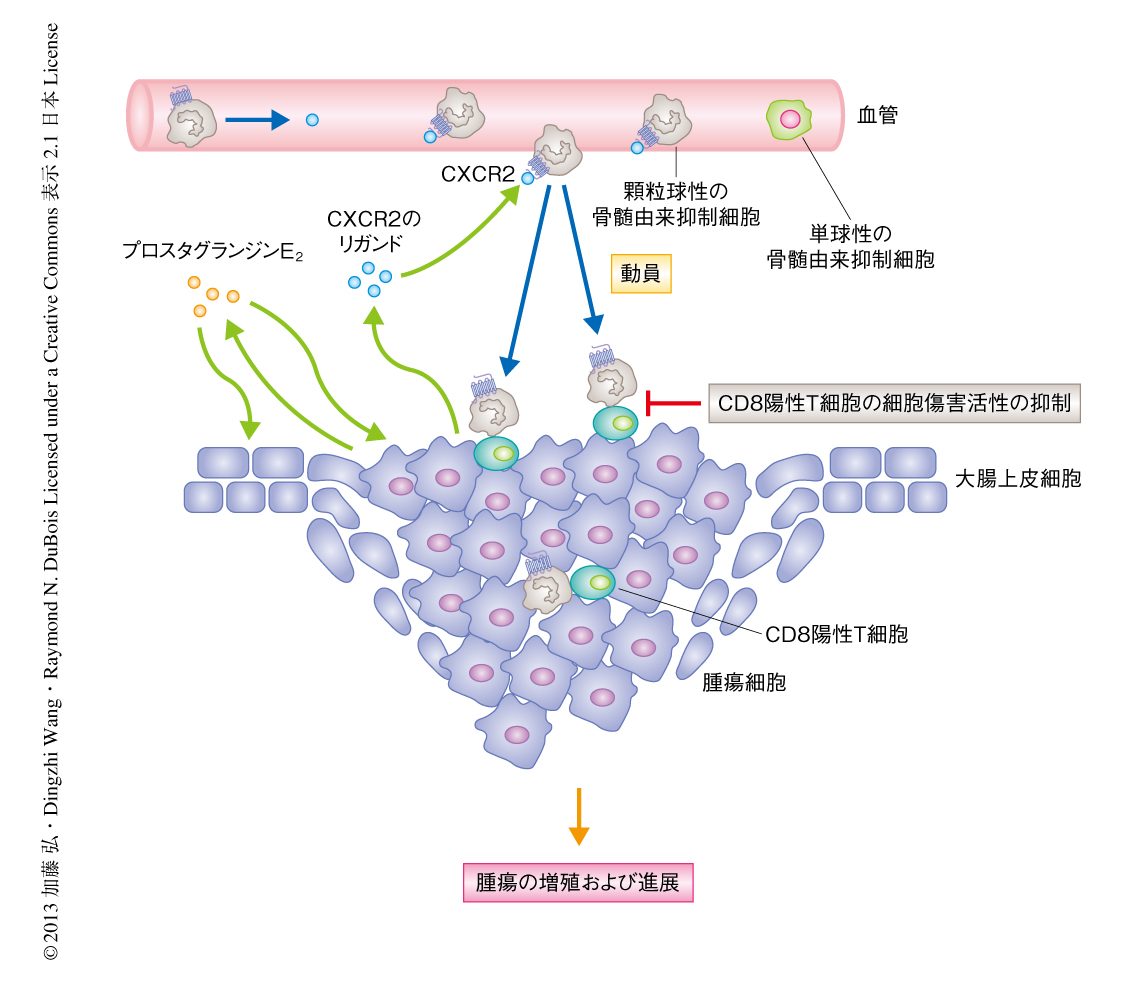
筆者らの研究室では,ヒトの大腸がん細胞および腫瘍異種移植片モデルにおいて炎症性メディエーターであるプロスタグランジンE2がケモカインCXCL1の発現を直接に促進することを報告したが14),大腸炎モデルマウスや炎症関連大腸腫瘍モデルマウス,あるいは,家族性大腸ポリポーシスや孤発性大腸がんの前悪性モデルとして広く使用されるApcmin/+マウスにおいて,プロスタグランジンE2がCXCR2のリガンドとなるケモカインの発現を制御しているかどうかは明らかでなかった.実際に,プロスタグランジンE2はデキストラン硫酸ナトリウムにより誘導した慢性大腸炎の組織,および,アゾキシメタンおよびデキストラン硫酸ナトリウムにより誘導した大腸腫瘍においてCXCL1およびCXCL2の産生を促進した.同様に,プロスタグランジンE2はApcmin/+マウスの腫瘍においてCXCL1およびCXCL2の産生を促進した.さらに,対照となるApcmin/+マウスの正常な粘膜においてもこれらのケモカインの産生を促進させた.一方,Apcmin/+マウスに対しシクロオキシゲナーゼ2に選択的な阻害剤を投与すると,CXCL1およびCXCL2の発現は完全に抑制された.したがって,プロスタグランジンE2などの炎症性メディエーターはCXCR2のリガンドとなるケモカインの発現を誘導することにより,CXCR2を発現する骨髄由来抑制細胞の遊走を促進することが示唆された(図2).
骨髄由来抑制細胞の浸潤について炎症関連大腸腫瘍の形成における直接的な機能を調べるため,野生型マウスから作製した炎症関連大腸腫瘍モデルマウスより単離した骨髄由来抑制細胞をCXCR2ノックアウトマウスに移入したところ,骨髄由来抑制細胞の大腸腫瘍組織への遊走が促進され,野生型の骨髄由来抑制細胞を移入しなかった場合,および,CXCR2を欠損した骨髄由来抑制細胞を移入した場合に比べ,大腸腫瘍の形成は有意に促進された.また,野生型の骨髄由来抑制細胞の移入により腫瘍における腺がんの比率も上昇した.以上から,ケモカイン受容体CXCR2を発現する骨髄由来抑制細胞は炎症関連大腸腫瘍の形成および進展に重要な役割をはたすと考えられた.
炎症関連大腸腫瘍モデルマウスを用いて,腫瘍免疫において中心的な役割をもつ細胞傷害性T細胞における骨髄由来抑制細胞の機能を解析した.野生型マウスとCXCR2ノックアウトマウスの両方において,細胞傷害性CD8陽性T細胞は周囲の正常な組織に比べ腫瘍組織において減少していたが,CXCR2の欠損は細胞傷害性T細胞の腫瘍組織における数には変化をもたらさなかった.一方,CXCR2ノックアウトマウスの大腸に由来する細胞傷害性T細胞は野生型マウスに由来する細胞傷害性T細胞よりも,アゾキシメタンおよびデキストラン硫酸ナトリウムにより誘導した腫瘍細胞に対し高い細胞傷害活性を示した.さらに,CXCR2ノックアウトマウスの大腸に由来する細胞傷害性CD8陽性T細胞においては活性化のマーカーであるCD107aの発現が亢進していた.CXCR2ノックアウトマウスの大腸腫瘍に由来する細胞傷害性CD8陽性T細胞において,細胞傷害活性の中心を担うインターフェロンγ,パーフォリン,グランザイムの発現は,野生型マウスに由来するCD8陽性T細胞よりも高かった.また,野生型マウスおよびCXCR2ノックアウトマウスの両方において,血中に由来する顆粒球性の骨髄由来抑制細胞はCD8陽性T細胞の細胞傷害活性やインターフェロンγおよびインターロイキン2の産生を細胞の数に依存して抑制した(図2).アゾキシメタンおよびデキストラン硫酸ナトリウムの投与により,野生型マウスおよびCXCR2ノックアウトマウスの両方の骨髄由来抑制細胞において,骨髄由来抑制細胞の活性化物質であるアルギナーゼ1の発現やアルギナーゼ活性の亢進がみられた.同様に,骨髄由来抑制細胞の免疫抑制活性の指標であるCD80およびCD86の発現も,野生型マウスおよびCXCR2ノックアウトマウスの両方の骨髄由来抑制細胞においてみられた.これらの結果より,大腸の組織に遊走した骨髄由来抑制細胞によるCD8陽性T細胞の抗腫瘍細胞としての機能は,ケモカイン受容体CXCR2の発現とは独立していることが考えられた.また,CXCR2の欠損により大腸腫瘍組織におけるTh1細胞の増加とTh17細胞の減少がみられた.したがって,CXCR2シグナル伝達系はTh1細胞とTh2細胞との不均衡,および,Th17細胞による抗腫瘍炎症反応を惹起する可能性があり,さらなる詳細な研究が必要である.
ケモカイン受容体CXCR2は顆粒球性の骨髄由来抑制細胞が大腸炎の組織および腫瘍組織に遊走するために必要であり,炎症に関連する大腸腫瘍の形成および進展に重要な役割をもつことが明らかにされた.さらに,炎症性メディエーターであるプロスタグランジンE2はCXCR2のリガンドとなるケモカインの大腸の粘膜および腫瘍細胞からの産生を促進することが示された.この研究の結果は,炎症性の腫瘍微小環境が腫瘍細胞の免疫監視からの逃避をひき起こす分子機構の解明に貢献するものと考えられる.また,腫瘍の免疫寛容を標的とした治療におけるCXCR2の阻害剤や中和抗体の可能性を示した.
略歴:2010年 北里大学大学院医療系研究科 修了,同年 同 助教,2011年 米国Texas大学MD Anderson Cancer CenterにてPostdoctoral Fellowを経て,2012年より米国Arizona州立大学Biodesign InstituteにてPostdoctoral Fellow.
研究テーマ: 腫瘍微小環境.
抱負:帰国ののち,トランスレーショナルリサーチの発展に寄与したい.
Dingzhi Wang
米国Arizona州立大学Biodesign InstituteにてResearch Professor.
Raymond N. DuBois
米国Arizona州立大学Biodesign InstituteにてExecutive Director.
© 2013 加藤 弘・Dingzhi Wang・Raymond N. DuBois Licensed under CC 表示 2.1 日本
(米国Arizona州立大学Biodesign Institute,Laboratory for Inflammation and Cancer)
email:加藤 弘
DOI: 10.7875/first.author.2013.147
CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis.
Hiroshi Katoh, Dingzhi Wang, Takiko Daikoku, Haiyan Sun, Sudhansu K. Dey, Raymond N. DuBois
Cancer Cell, 24, 631-644 (2013)
この論文に出現する遺伝子・タンパク質のUniprot ID
ケモカイン受容体CXCR2(P35343), CXCR2(P35343), ケモカイン受容体, ケモカイン, CD8, インターロイキン10(P18893), シクロオキシゲナーゼ2(P35355), 炎症性ケモカイン, ケモカインCXCL1(P12850), CXCL1(P12850), CD11b(P05555), Gr-1, Ly6G(P35461), Ly6C, CXCL2(P10889), CXCL5(P50228), インターロイキン6(P05231), Stat3(P40763), Apc(Q61315), CD107a(P11438), インターフェロンγ(P01580), パーフォリン(P10820), グランザイム, インターロイキン2(P04351), アルギナーゼ1(Q61176), アルギナーゼ, CD80(Q00609), CD86(P42082), CXCR1(P25024)
要 約
炎症性腸疾患などの慢性炎症は発がんおよび腫瘍の進展におけるリスク因子のひとつである.しかしながら,炎症および炎症性メディエーターの発がんにおける詳細な分子機構はいまだ解明されていない.この研究では,ケモカイン受容体であるCXCR2をノックアウトしたマウスを用いた大腸炎に関連する腫瘍のモデルマウスを用いて,CXCR2の発現が骨髄由来抑制細胞の炎症の局所への遊走に不可欠であることを明らかにした.CXCR2のリガンドとなるケモカインの産生は炎症を生じた大腸の粘膜および腫瘍において亢進しており,骨髄由来抑制細胞の局所への浸潤を促進していた.野生型の骨髄由来抑制細胞をCXCR2ノックアウトマウスへ移入することにより,炎症に関連する腫瘍の形成および進展は回復することが示された.また,局所に浸潤した骨髄由来抑制細胞はCD8陽性T細胞の細胞傷害活性を抑制することにより,腫瘍細胞の免疫回避を誘導していた.これらの発見が,炎症に関連する大腸腫瘍の進展の分子機構の解明にくわえ,腫瘍において免疫寛容を標的とする新規の診断法あるいは治療法につながることが期待される.
はじめに
大腸がんは悪性腫瘍のうち世界第2位の罹患率,第4位の死亡数をもたらしているが,大腸内視鏡検査の普及にもかかわらず,いまだ半数以上の症例は初診のとき進行がんとしてみつかっている1).遺伝子の変異あるいはエピジェネティックな変異にくわえて,慢性炎症は大腸がんのリスク因子のひとつである2).実際に,潰瘍性大腸炎の20%以上の症例は診断ののち30年以内に大腸がんを発生することが報告されている3).このように慢性炎症と大腸がんには密接な関連がみられることから,大腸炎に関連する腫瘍の研究は“proof of concept”モデルとして,慢性炎症および炎症性メディエーターの腫瘍の発生,進展,転移における分子機構の解明につながると考えられる.
慢性炎症は損傷や病原微生物への暴露によりもたらされる免疫反応の亢進状態が持続することにより生じる.実際に,腸内細菌叢の重要性は炎症性腸疾患の患者において証明されている4).慢性大腸炎が腸内細菌叢の存在に依存していることは動物実験により直接的に示唆され5),インターロイキン10ノックアウトマウスを使用した炎症関連大腸がんモデルマウスは無菌状態では大腸がんを発生しない6).同様に,遺伝性および孤発性の大腸がんモデルマウスでも腸内細菌叢が腫瘍の増殖にかかわることが示唆されている7).注目すべきことに,病原細菌は炎症を起こした大腸の粘膜においてシクロオキシゲナーゼ2の発現を誘導し8),シクロオキシゲナーゼ2とそれに由来する炎症性メディエーターであるプロスタグランジンE2は炎症性腸疾患の症例の大腸の粘膜においていちじるしく増加している9).
炎症性腸疾患において,炎症性細胞の著明な浸潤は主要な組織学的な特徴のひとつである.同様に,炎症に関連する大腸がんおよび孤発性の大腸がんにおいてもさまざまな免疫細胞や内皮細胞が動員され,腫瘍微小環境を構築する10).ケモカインは循環血から炎症の局所あるいは腫瘍に炎症性細胞を動員するため不可欠である.腸の粘膜における炎症性ケモカインの増加と炎症性細胞の著明な浸潤は炎症性腸疾患において疾患の程度と密接に相関する.炎症性ケモカインは孤発性大腸がんの大腸上皮においてもいちじるしく亢進している11).しかしながら,こうしたケモカインおよびケモカイン受容体の炎症性腸疾患および大腸がんにおける詳細な分子機構はいまだ明らかになっていない.
がんの発生と進展は宿主の免疫監視からの腫瘍細胞の逃避にも依存している.大腸がんなど固形がんにおいては,Th1細胞とTh2細胞との不均衡や,骨髄由来抑制細胞(myeloid-derived suppressor cell:MDSC)あるいは制御性T細胞など免疫抑制細胞の亢進が生じている.血中の骨髄由来抑制細胞の数が大腸がんの臨床病期や遠隔転移と相関することが報告されている12).骨髄由来抑制細胞はT細胞やナチュラルキラー細胞の機能を抑制することによりがん細胞の免疫逃避に貢献する13).しかしながら,骨髄由来抑制細胞の循環血から局所への動員の分子機構や大腸がんの進展にかかわる詳細な機序についてはいまだ不明であった.
筆者らの研究室では,炎症性メディエーターであるプロスタグランジンE2が大腸がん細胞からのケモカインCXCL1の産生を直接に促進し,それにより腫瘍における血管の新生が誘導されることを報告した14).CXCL1はケモカイン受容体CXCR2のリガンドのひとつであり(図1a),CXCR2は好中球に発現しその炎症性の動員において中心的な役割を担うことが知られている15).炎症性腸疾患において血清のCXCL1の量は増加しており,疾患の程度と相関する16).同様に,CXCL1およびCXCR2の発現が孤発性大腸がんの組織において上昇していることを報告した14).この研究では,炎症に関連する大腸腫瘍におけるCXCR2シグナル伝達系の分子機構について検討した.
1.ケモカイン受容体CXCR2のノックアウトは炎症に関連する大腸腫瘍の発生を抑制する
炎症に関連する大腸腫瘍の発症におけるCXCR2シグナル伝達系の役割を調べるため,野生型マウスおよびCXCR2ノックアウトマウスに大腸に特異的な発がん物質であるアゾキシメタンを腹腔内投与し,ひきつづき,大腸炎をひき起こす薬剤であるデキストラン硫酸ナトリウムを4サイクルにわたり服用させる,慢性大腸炎および炎症関連大腸腫瘍モデルマウスを使用した.この2つの薬剤の投与により,野生型マウスにおいては血便および大腸の短縮などの肉眼所見がみられ,組織的な炎症スコアの有意な上昇が認められた.CXCR2ノックアウトマウスではこれらの肉眼所見および組織的な炎症の発生は有意に抑制されていた.さらに,CXCR2ノックアウトマウスでは腫瘍細胞の増殖の抑制による炎症関連大腸腫瘍の劇的な減少が認められた.炎症の程度と腫瘍の数は明らかに相関していた.また,CXCR2ノックアウトマウスでは腫瘍における腺がんの比率も低下しており,CXCR2は腫瘍の進展を促進すると考えられた.以上から,ケモカイン受容体CXCR2は慢性大腸炎および炎症関連腫瘍の形成および進展に重要な役割をもつことが示唆された.
2.ケモカイン受容体CXCR2のノックアウトにより循環血における骨髄由来抑制細胞の大腸炎あるいは腫瘍への動員は抑制される
CXCR2が免疫細胞の動員に関与しているかどうかを調べるため,まず,デキストラン硫酸ナトリウムにより誘導した大腸炎モデルマウスにおける大腸炎症の組織において,免疫細胞のプロファイルを解析した.デキストラン硫酸ナトリウムの単回投与による急性炎症期ではさまざまな免疫細胞が局所に動員され,CXCR2の欠損により好中球,マクロファージ,骨髄由来抑制細胞の動員が抑制された.一方,デキストラン硫酸ナトリウムのくり返し投与による慢性炎症期ではCXCR2の欠損により骨髄由来抑制細胞の動員のみが有意に抑制された.
マウスでは,未熟な骨髄に由来する細胞(しばしば,骨髄由来抑制細胞)はCD11b陽性Gr-1陽性細胞として定義され,さらに,顆粒球性の骨髄由来抑制細胞(CD11b陽性Ly6G陽性細胞)と単球性の骨髄由来抑制細胞(CD11b陽性Ly6C陽性細胞)とに分けられる17)(図1b).大腸炎の組織では90%以上の骨髄由来抑制細胞が顆粒球性であった.興味深いことに,CXCR2ノックアウトマウスでは正常な状態および大腸炎の両方において循環血に顆粒球性の骨髄由来抑制細胞の蓄積が認められ,これは慢性炎症によりさらに蓄積した.このことから,CXCR2を欠損した骨髄由来抑制細胞は局所への浸潤能が欠損しているものと考えられた.注目すべきことに,血中の顆粒球性の骨髄由来抑制細胞の大部分はCXCR2を発現していたが,単球性の骨髄由来抑制細胞にはCXCR2の発現は認められなかった.慢性大腸炎モデルマウスと同様に,アゾキシメタンおよびデキストラン硫酸ナトリウムにより誘導した炎症関連大腸腫瘍においても顆粒球性の骨髄由来抑制細胞の大腸の組織への浸潤の亢進が認められ,腫瘍組織では周囲の炎症組織よりもさらに高度な浸潤がみられた.CXCR2の欠損により顆粒球性の骨髄由来抑制細胞の腫瘍組織および炎症組織の両方への動員は著明に抑制され,慢性炎症と同様の免疫細胞のプロファイルを示した.これを反映し,CXCR2ノックアウトマウスの血中には顆粒球性の骨髄由来抑制細胞の蓄積が認められた.一方,CXCR2ノックアウトマウスの循環血における顆粒球性の骨髄由来抑制細胞の生存率は野生型マウスより有意に低下していた.以上から,ケモカイン受容体CXCR2は顆粒球性の骨髄由来抑制細胞の循環血より炎症組織および腫瘍への動員に不可欠であることが示された.
3.ケモカイン受容体CXCR2はそのリガンドにより誘導される顆粒球性の骨髄由来抑制細胞の化学遊走を介在する
以上の結果から,CXCR2を発現する免疫細胞の動員に不可欠であるCXCR2のリガンドとなるケモカインの発現は炎症組織および腫瘍組織において上昇していると予想された.実際に,野生型マウスでは大腸炎組織においてケモカインCXCL1,CXCL2,CXCL5の発現が上昇しており,腫瘍組織ではさらにこれらの発現が亢進していた.組織におけるCXCR2のリガンドとなるケモカインの発現と顆粒球性の骨髄由来抑制細胞の浸潤の程度とは相関していた.CXCR2のリガンドとなるケモカインを産生する細胞を同定するためin situハイブリダイゼーションを行ったところ,これらのmRNAはおもに大腸上皮,とくに,腫瘍細胞において発現していた.
CXCR2のリガンドとなるケモカインによる細胞遊走アッセイを行ったところ,野生型の顆粒球性の骨髄由来抑制細胞は遊走を示したがCXCR2を欠損した顆粒球性の骨髄由来抑制細胞には遊走が認められなかったことから,CXCR2がリガンドとなるケモカインに誘導される化学遊走に必須であることが証明された.
骨髄に由来する細胞はインターロイキン6の主要な産生細胞であり,インターロイキン6-Stat3シグナル伝達系は骨髄由来抑制細胞の免疫抑制機能において中心的な役割をはたす13).用いたモデルマウスにおいてインターロイキン6の発現は大腸炎を生じた粘膜および腫瘍組織において上昇しており,CXCR2ノックアウトマウスでは抑制されていた.血清におけるインターロイキン6の発現はCXCR2ノックアウトマウスにおいて亢進しており,骨髄由来抑制細胞と同様のパターンを示した.また,リン酸化Stat3の発現は野生型マウスの腫瘍間質細胞には認められたが,CXCR2ノックアウトマウスには認められなかった.したがって,大腸組織に浸潤した骨髄由来抑制細胞は免疫抑制作用をもつ可能性が示唆された.以上から,CXCR2リガンド-CXCR2シグナル伝達系は炎症組織および腫瘍組織への動員において必須であることが証明された(図2).
4.炎症性メディエーターであるプロスタグランジンE2は大腸の粘膜および腫瘍におけるCXCR2のリガンドの発現を促進する
筆者らの研究室では,ヒトの大腸がん細胞および腫瘍異種移植片モデルにおいて炎症性メディエーターであるプロスタグランジンE2がケモカインCXCL1の発現を直接に促進することを報告したが14),大腸炎モデルマウスや炎症関連大腸腫瘍モデルマウス,あるいは,家族性大腸ポリポーシスや孤発性大腸がんの前悪性モデルとして広く使用されるApcmin/+マウスにおいて,プロスタグランジンE2がCXCR2のリガンドとなるケモカインの発現を制御しているかどうかは明らかでなかった.実際に,プロスタグランジンE2はデキストラン硫酸ナトリウムにより誘導した慢性大腸炎の組織,および,アゾキシメタンおよびデキストラン硫酸ナトリウムにより誘導した大腸腫瘍においてCXCL1およびCXCL2の産生を促進した.同様に,プロスタグランジンE2はApcmin/+マウスの腫瘍においてCXCL1およびCXCL2の産生を促進した.さらに,対照となるApcmin/+マウスの正常な粘膜においてもこれらのケモカインの産生を促進させた.一方,Apcmin/+マウスに対しシクロオキシゲナーゼ2に選択的な阻害剤を投与すると,CXCL1およびCXCL2の発現は完全に抑制された.したがって,プロスタグランジンE2などの炎症性メディエーターはCXCR2のリガンドとなるケモカインの発現を誘導することにより,CXCR2を発現する骨髄由来抑制細胞の遊走を促進することが示唆された(図2).
5.CXCR2ノックアウトマウスへ野生型の骨髄由来抑制細胞を移入することにより腫瘍の形成は促進される
骨髄由来抑制細胞の浸潤について炎症関連大腸腫瘍の形成における直接的な機能を調べるため,野生型マウスから作製した炎症関連大腸腫瘍モデルマウスより単離した骨髄由来抑制細胞をCXCR2ノックアウトマウスに移入したところ,骨髄由来抑制細胞の大腸腫瘍組織への遊走が促進され,野生型の骨髄由来抑制細胞を移入しなかった場合,および,CXCR2を欠損した骨髄由来抑制細胞を移入した場合に比べ,大腸腫瘍の形成は有意に促進された.また,野生型の骨髄由来抑制細胞の移入により腫瘍における腺がんの比率も上昇した.以上から,ケモカイン受容体CXCR2を発現する骨髄由来抑制細胞は炎症関連大腸腫瘍の形成および進展に重要な役割をはたすと考えられた.
6.大腸の腫瘍組織に遊走した骨髄由来抑制細胞はCD8陽性T細胞の細胞傷害活性を抑制する
炎症関連大腸腫瘍モデルマウスを用いて,腫瘍免疫において中心的な役割をもつ細胞傷害性T細胞における骨髄由来抑制細胞の機能を解析した.野生型マウスとCXCR2ノックアウトマウスの両方において,細胞傷害性CD8陽性T細胞は周囲の正常な組織に比べ腫瘍組織において減少していたが,CXCR2の欠損は細胞傷害性T細胞の腫瘍組織における数には変化をもたらさなかった.一方,CXCR2ノックアウトマウスの大腸に由来する細胞傷害性T細胞は野生型マウスに由来する細胞傷害性T細胞よりも,アゾキシメタンおよびデキストラン硫酸ナトリウムにより誘導した腫瘍細胞に対し高い細胞傷害活性を示した.さらに,CXCR2ノックアウトマウスの大腸に由来する細胞傷害性CD8陽性T細胞においては活性化のマーカーであるCD107aの発現が亢進していた.CXCR2ノックアウトマウスの大腸腫瘍に由来する細胞傷害性CD8陽性T細胞において,細胞傷害活性の中心を担うインターフェロンγ,パーフォリン,グランザイムの発現は,野生型マウスに由来するCD8陽性T細胞よりも高かった.また,野生型マウスおよびCXCR2ノックアウトマウスの両方において,血中に由来する顆粒球性の骨髄由来抑制細胞はCD8陽性T細胞の細胞傷害活性やインターフェロンγおよびインターロイキン2の産生を細胞の数に依存して抑制した(図2).アゾキシメタンおよびデキストラン硫酸ナトリウムの投与により,野生型マウスおよびCXCR2ノックアウトマウスの両方の骨髄由来抑制細胞において,骨髄由来抑制細胞の活性化物質であるアルギナーゼ1の発現やアルギナーゼ活性の亢進がみられた.同様に,骨髄由来抑制細胞の免疫抑制活性の指標であるCD80およびCD86の発現も,野生型マウスおよびCXCR2ノックアウトマウスの両方の骨髄由来抑制細胞においてみられた.これらの結果より,大腸の組織に遊走した骨髄由来抑制細胞によるCD8陽性T細胞の抗腫瘍細胞としての機能は,ケモカイン受容体CXCR2の発現とは独立していることが考えられた.また,CXCR2の欠損により大腸腫瘍組織におけるTh1細胞の増加とTh17細胞の減少がみられた.したがって,CXCR2シグナル伝達系はTh1細胞とTh2細胞との不均衡,および,Th17細胞による抗腫瘍炎症反応を惹起する可能性があり,さらなる詳細な研究が必要である.
おわりに
ケモカイン受容体CXCR2は顆粒球性の骨髄由来抑制細胞が大腸炎の組織および腫瘍組織に遊走するために必要であり,炎症に関連する大腸腫瘍の形成および進展に重要な役割をもつことが明らかにされた.さらに,炎症性メディエーターであるプロスタグランジンE2はCXCR2のリガンドとなるケモカインの大腸の粘膜および腫瘍細胞からの産生を促進することが示された.この研究の結果は,炎症性の腫瘍微小環境が腫瘍細胞の免疫監視からの逃避をひき起こす分子機構の解明に貢献するものと考えられる.また,腫瘍の免疫寛容を標的とした治療におけるCXCR2の阻害剤や中和抗体の可能性を示した.
文 献
- Yamashita, K. & Watanabe, M.: Clinical significance of tumor markers and an emerging perspective on colorectal cancer. Cancer Sci., 100, 195-199 (2009)[PubMed]
- Ekbom, A., Helmick, C., Zack, M. et al.: Ulcerative colitis and colorectal cancer. A population-based study. N. Engl. J. Med., 323, 1228-1233 (1990)[PubMed]
- Lakatos, P. L. & Lakatos, L.: Risk for colorectal cancer in ulcerative colitis: changes, causes and management strategies. World J. Gastroenterol., 14, 3937-3947 (2008)[PubMed]
- Gionchetti, P., Rizzello, F., Helwig, U. et al. .: Prophylaxis of pouchitis onset with probiotic therapy: a double-blind, placebo-controlled trial. Gastroenterology, 124, 1202-1209 (2003)[PubMed]
- Elson, C. O., Cong, Y., McCracken, V. J. et al.: Experimental models of inflammatory bowel disease reveal innate, adaptive, and regulatory mechanisms of host dialogue with the microbiota. Immunol. Rev., 206, 260-276 (2005)[PubMed]
- Uronis, J. M., Muhlbauer, M., Herfarth, H. H. et al.: Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS One, 4, e6026 (2009)[PubMed]
- Grivennikov, S., Karin, E., Terzic, J. et al.: IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell, 15, 103-113 (2009)[PubMed]
- Abdallah Hajj Hussein, I., Freund, J. N., Reimund, J. M. et al.: Enteropathogenic e.coli sustains iodoacetamide-induced ulcerative colitis-like colitis in rats: modulation of IL-1beta, IL-6, TNF-alpha, COX-2, and apoptosisi. J. Biol. Regul. Homeost. Agents, 26, 515-526 (2012)[PubMed]
- Singer, I. I., Kawka, D. W., Schloemann, S. et al.: Cyclooxygenase 2 is induced in colonic epithelial cells in inflammatory bowel disease. Gastroenterology, 115, 297-306 (1998)[PubMed]
- Coussens, L. M. & Werb, Z.: Inflammation and cancer. Nature, 420, 860-867 (2002)[PubMed]
- Fegn, L. & Wang, Z.: Topical chemoprevention of skin cancer in mice, using combined inhibitors of 5-lipoxygenase and cyclo-oxygenase-2. J. Laryngol. Otol., 123, 880-884 (2009)[PubMed]
- Diaz-Montero, C. M., Salem, M. L., Nishimura, M. I. et al.: Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother., 58, 49-59 (2009)[PubMed]
- Gabrilovich, D. I. & Nagaraj, S.: Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol., 9, 162-174 (2009)[PubMed]
- Wang, D., Wang, H., Brown, J. et al.: CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer. J. Exp. Med., 203, 941-951 (2006)[PubMed]
- Oppenheim, J. J., Zachariae, C. O., Mukaida, N. et al.: Properties of the novel proinflammatory supergene "intercrine" cytokine family. Annu. Rev. Immunol., 9, 617-648 (1991)[PubMed]
- Mitsuyama, K., Tsuruta, O., Tomiyasu, N. et al.: Increased circulating concentrations of growth-related oncogene (GRO)-α in patients with inflammatory bowel disease. Dig. Dis. Sci., 51, 173-177 (2006)[PubMed]
- Youn, J. I., Nagaraj, S., Collazo, M. et al.: Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol., 181, 5791-5802 (2008)[PubMed]
著者プロフィール
略歴:2010年 北里大学大学院医療系研究科 修了,同年 同 助教,2011年 米国Texas大学MD Anderson Cancer CenterにてPostdoctoral Fellowを経て,2012年より米国Arizona州立大学Biodesign InstituteにてPostdoctoral Fellow.
研究テーマ: 腫瘍微小環境.
抱負:帰国ののち,トランスレーショナルリサーチの発展に寄与したい.
Dingzhi Wang
米国Arizona州立大学Biodesign InstituteにてResearch Professor.
Raymond N. DuBois
米国Arizona州立大学Biodesign InstituteにてExecutive Director.
© 2013 加藤 弘・Dingzhi Wang・Raymond N. DuBois Licensed under CC 表示 2.1 日本
