アポトーシスを誘導するキナーゼMst1はBeclin1とBcl-2との結合を促進することによりオートファジーを抑制する
前嶋康浩・佐渡島純一
(米国Rutgers New Jersey Medical School,Department of Cell Biology and Molecular Medicine)
email:前嶋康浩,佐渡島純一
DOI: 10.7875/first.author.2013.138
Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2.
Yasuhiro Maejima, Shiori Kyoi, Peiyong Zhai, Tong Liu, Hong Li, Andreas Ivessa, Sebastiano Sciarretta, Dominic P Del Re, Daniela K Zablocki, Chiao-Po Hsu, Dae-Sik Lim, Mitsuaki Isobe, Junichi Sadoshima
Nature Medicine, 19, 1478-1488 (2013)
筆者らは,アポトーシスを誘導するキナーゼであるMst1に,オートファジーを阻害してタンパク質の品質管理システム機構を負に制御する機能があることを見い出した.ストレスにより活性化されたMst1は心筋細胞においてオートファゴソームの形成を抑制し,p62の集積とアグリソームの蓄積を促進した.Mst1はBeclin1のBH3ドメインにあるThrをリン酸化し,Beclin1とBcl-2あるいはBcl-xLとの結合を強化してBeclin1ホモ二量体を安定化させた.その結果,Vps34複合体IのPI3キナーゼ活性が抑制されオートファゴソームの形成が阻害された.さらに,Mst1がBeclin1とBcl-2あるいはBcl-xLとの結合を促進することにより,アポトーシス促進タンパク質であるBaxのBcl-2あるいはBcl-xLからの解離が促進されアポトーシスが誘導された.心筋梗塞や拡張型心筋症における不全な心筋においてはMst1の活性が上昇し,オートファジーが抑制されアグリソームの蓄積が顕著であった.これらの結果は,Mst1がBeclin1をリン酸化してBeclin1,Bcl-2あるいはBcl-xL,Baxとの結合能を制御することにより,オートファジーとアポトーシスとを協調的に制御していることを示唆した.
オートファジーは変性したタンパク質や障害をうけたオルガネラを除去して細胞における恒常性を維持する役割を担っている.Atg5遺伝子の欠損によりオートファジーの抑制されたマウスの心筋ではタンパク質の凝集が蓄積して心臓の機能が低下することから,オートファジーは心臓の機能の維持に欠かせないことが示唆されている1).また,ストレスにより活性化するオートファジーは心筋に対し保護的に作用することが一般的であるが2),その一方で,虚血後の再灌流のときのようにオートファジーが過剰に活性化すると心筋細胞はダメージをうける3).このように,オートファジーが生理的なレベルより抑制されること,あるいは,過剰に活性化することは,心筋細胞にとり有害である.しかし,オートファジーを最適なレベルに保つ分子機序については,いまだ解明されていない.
オートファジーの進展に重要な役割を担うタンパク質であるBeclin1は,クラスIIIのPI3キナーゼであるVps34と結合して2つの異なる複合体を形成する4).そのうち,Beclin1,Vps34,Atg14Lから構成されるVps34複合体Iは初期のオートファゴソームの形成に関与している.また,Bcl-2ファミリータンパク質であるBcl-2あるいはBcl-xLはBeclin1と結合してBeclin1-Vps34複合体の形成を阻害する5).しかし,Beclin1とBcl-2あるいはBcl-xLとの相互作用がどのような機序によりBeclin1のはたらきを抑制しているのかについては不明である.
Hippoシグナル伝達経路の主要な制御タンパク質であるセリン/スレオニンキナーゼMst1は,心筋細胞にアポトーシスが誘導されたときもっとも活性化しているキナーゼのひとつである6,7).筆者らは,これまでに,心筋においてMst1を恒常的に活性化させると拡張型心筋症が発症することや,内在性のMst1を阻害するとストレスにより誘導される心筋細胞のアポトーシスが抑制されることを見い出してきた6,8).さらに,この研究において,心筋においてMst1を恒常的に活性化させると,心筋細胞においてアグリソームやオートファジーの代表的な基質であるp62が著明に蓄積することも見い出した.
筆者らは,以上の背景より,Mst1が活性化するとオートファジーを抑制して心筋細胞に悪影響を及ぼし,心不全の発症を促すのではないかと考えた.
筆者らは以前に,Mst1には心筋梗塞ののち左室の機能不全を進行させる作用のあることを報告した8).心不全を起こした心筋にはしばしばアグリソームの蓄積を認めるが,陳旧性心筋梗塞モデルにおける梗塞境界領域の心筋細胞にもアグリソームが著明に蓄積していた.一方,Mst1ノックアウトマウスおよび心筋に特異的にドミナントネガティブ型のMst1を過剰に発現させたトランスジェニックマウスでは,心筋梗塞ののちの心筋では,野生型マウスの心筋梗塞ののちの心筋と比べアグリソームの蓄積が著しく減少していた.また,野生型マウスの心筋梗塞ののちの心筋では,偽手術を行った心筋と比較してオートファゴソームの数が増加していたが,Mst1ノックアウトマウスの心筋梗塞ののちの心筋では,野生型マウスの心筋梗塞ののちの心筋と比較してオートファゴソームの数はさらに増加していた.これらの結果と,心筋細胞に蓄積していたアグリソームにp62が共局在していたことを考えあわせると,Mst1がオートファジーを抑制するために心筋梗塞ののちの心筋においてアグリソームが蓄積してしまうのではないかと考えた.この仮説を検証するため,Mst1ノックアウトマウスとBeclin1ヘテロノックアウトマウスとを交配させオートファジーを阻害したところ,心筋におけるアグリソームの蓄積が増加した.また,Mst1ノックアウトマウスでみられた心筋梗塞ののちの左室のリモデリングや左室の収縮能および生存率の改善の効果は,Mst1ノックアウトマウスとBeclin1ヘテロノックアウトマウスとを交配したマウスでは認められなかった.以上より,Mst1の阻害により心筋梗塞ののちに心臓の機能が改善する機序には,オートファジーの制御が関与していることが示唆された.
Mst1の活性化がアグリソームの形成に関与しているかどうかにつき検証した.Mst1を心筋に特異的に過剰に発現させたトランスジェニックマウスの心筋では,ポリユビキチン化タンパク質やp62が共局在するアグリソームの蓄積が増加していた.また,培養心筋細胞においてMst1はオートファジーによる長寿命タンパク質の分解を抑制した.さらに,Mst1の心筋に特異的な過剰発現トランスジェニックマウスの心筋では心筋細胞におけるオートファゴソームの数が減少していたのに対し,Mst1ノックアウトマウスではオートファゴソームの数は増加していた.以上の結果から,Mst1にはオートファジーを抑制し変性タンパク質の蓄積を促進する作用のあることが示された.オートファゴソームの蛍光のマーカーであるmRFP-GFP-LC3をアデノウイルスを用いて発現させた培養心筋細胞において,Mst1はオートリソソームの数を減少させることが確認されたことから,Mst1はオートファジーフラックスを抑制することが示唆された.
Mst1によるオートファジーの制御機序につき検討した.Mst1がさまざまなオートファジー制御タンパク質と相互作用をするかどうかを免疫沈降法により検討したところ,Mst1はBeclin1と強い結合を示した.また,Mst1はBeclin1とVps34との結合,Beclin1とAtg14Lとの結合をいずれも抑制し,Vps34複合体Iのキナーゼ活性を抑制した.さらに,Mst1はBcl-2あるいはBcl-xLとBeclin1との結合を促進することも見い出した.Beclin1のBH3ドメインを模倣したABT-737は,Mst1のオートファジーの抑制作用を阻害し,Mst1によるBcl-2とBeclin1との結合の増強効果を抑制した.以上より,Mst1はBeclin1のBH3ドメインとBcl-2あるいはBcl-xLとの結合能を増強させることにより,オートファジーを抑制していることが示唆された.さらに,Mst1はBeclin1ホモ二量体の形成を促進し,Beclin1およびAtg14Lが構成するVps34複合体Iの形成を阻害していることも見い出した.
in vitroキナーゼアッセイにより,Mst1がBeclin1をリン酸化することが見い出された.興味深いことに,Mst1によるBeclin1のリン酸化の部位は,Bcl-2あるいはBcl-xLとの結合部位と想定されるBH3ドメインにあるThr108であることが質量分析法により判明した.実際に,内在性のMst1がBeclin1をリン酸化していることは,Thr108に特異的なリン酸化抗体を用いて確認された.また,この抗体を用いて,Mst1は小胞体に局在するBeclin1をリン酸化していることも見い出した.さらに,Mst1がBeclin1をリン酸化すると,Beclin1とBcl-2あるいはBcl-xLとの結合が強化されることも見い出した.このことは,Mst1によるリン酸化を模倣したBeclin1変異体がBcl-2あるいはBcl-xLと恒常的に強く結合すること,Mst1によりリン酸化されないBeclin1変異体ではBcl-2あるいはBcl-xLとの結合が抑制されることから確認された.Beclin1はDAPKによってもリン酸化されるが,Mst1とは逆に,DAPKはBeclin1とBcl-2あるいはBcl-xLとの解離を促進してオートファジーを誘導する.同様に,JNK1によるBcl-2あるいはBcl-xLのリン酸化もBeclin1とBcl-2あるいはBcl-xLとの解離を促進する.これらDAPKまたはJNK1によるリン酸化を介したオートファジーの活性化には,基底状態においてBeclin1とBcl-2あるいはBcl-xLとが結合している必要がある.Beclin1とBcl-2あるいはBcl-xLの結合を促進する役割を担うMst1は,Beclin1を介したオートファジーの制御に欠かせない生理的なキナーゼであると考えられた.
Mst1によるBeclin1のリン酸化がBeclin1とBcl-2あるいはBcl-xLの結合を促進する分子機構を構造生物学的な観点から検討した.Beclin1-Bcl-xL複合体の結晶構造解析によると9),Beclin1のThr108はBH3ドメインのN末端側に位置しており,Bcl-xLの形成する疎水性の溝の入口に位置するα3ヘリックスと結合する.変異タンパク質を用いたプルダウンアッセイにより,Thr108がリン酸化されて陰電荷をもつようになったBeclin1のBH3ドメインのN末端側は,Bcl-2のα3ヘリックスにある塩基性アミノ酸His117との結合が強固になり,Beclin1とBcl-xLとの結合が増強されることを見い出した.
Beclin1ホモ二量体の形成はコイルドコイルドメインを介して起こる.Beclin1ホモ二量体のコイルドコイルドメインにおける結合構造は不安定であるため,コイルドコイルドメインにおける結合構造の安定しているBeclin1-Atg14Lヘテロ二量体に遷移しやすい10).Bcl-2はBH3ドメインだけではなく,コイルドコイルドメインにおいてもBeclin1と結合すること,Beclin1のBH3ドメインとBcl-2との結合能はMst1によるリン酸化により増強されるが,Beclin1のコイルドコイルドメインはBcl-2のBH4ドメインと結合し,その結合能はMst1によるリン酸化の影響をうけないことを見い出した.このことは,Bcl-2にはMst1に依存して2つのBeclin1をつなぎとめ,Beclin1ホモ二量体を安定化させるはたらきのあることを示した.Mst1を抑制するとBeclin1とAtg14Lとの結合は強くなることから,Mst1によるBeclin1のリン酸化はBeclin1ホモ二量体を安定化させる一方で,Beclin1からのAtg14Lの解離を促進し,その結果,Vps34複合体Iの形成およびその活性化を阻害していると考えられた(図1).
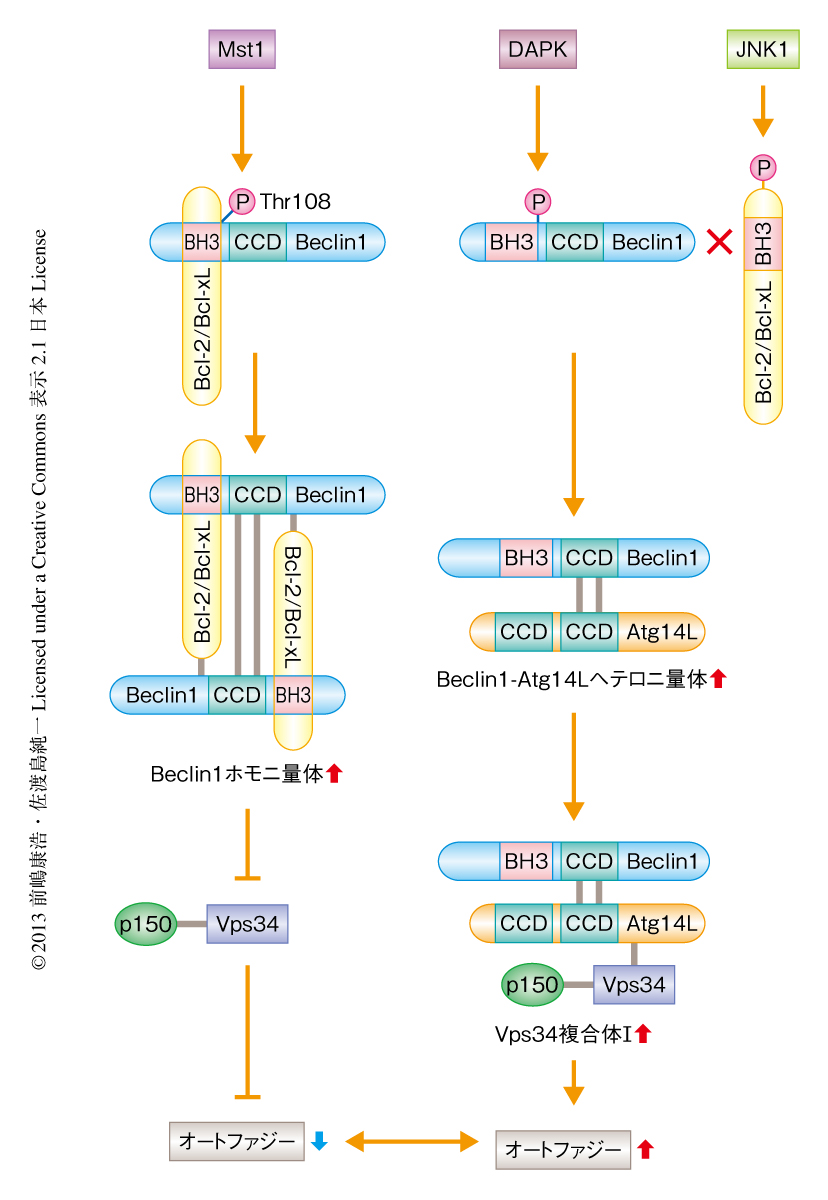
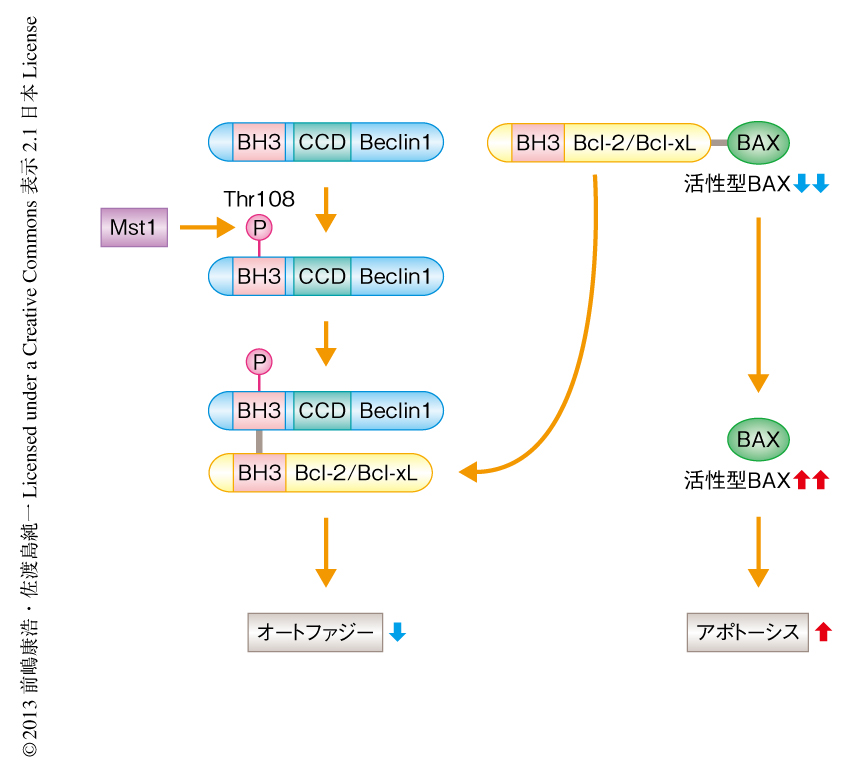
Mst1によるBeclin1のリン酸化がオートファジーに対し及ぼす影響について検討した.Mst1によるリン酸化を模倣したBeclin1変異体を導入した心筋細胞では,Mst1の有無にかかわらずVps34複合体Iのキナーゼ活性およびオートファジーが著明に抑制された一方,Mst1によりリン酸化されないBeclin1変異体を導入した心筋細胞では,Mst1によるVps34複合体Iのキナーゼ活性の抑制は認められず,Mst1を過剰に発現させてもオートファジーの抑制はみられなかった.これらの結果より,Mst1によるBeclin1のリン酸化はオートファジーの抑制に重要なはたらきをもつことが示唆された.Mst1を心筋細胞に過剰に発現させると,Beclin1とBcl-2との結合を促進するだけでなく,Bcl-2とアポトーシス促進タンパク質であるBaxとの結合能が低下し,Baxは活性化した.Mst1によるリン酸化を模倣したBeclin1変異体を心筋細胞に導入すると,Mst1の有無にかかわらず心筋細胞のアポトーシスが促進される一方,Mst1によりリン酸化されないBeclin変異体を心筋細胞に導入すると,Mst1により誘導されるアポトーシスは抑制された.このように,Mst1により促進されるBeclin1とBcl-2との結合の増強作用は,心筋細胞においてオートファジーを抑制するだけでなく,Bcl-2からのBaxの解離および活性化を促進することにより,アポトーシスを促進するはたらきのあることが示唆された(図2).
6週間が経過した心筋梗塞モデルマウスの心筋においてThr108のリン酸化されたBeclin1が増加していたが,Mst1ノックアウトマウスの心筋においては増加していなかったことから,心筋梗塞ののちの心筋において,内在性のMst1はBeclin1をリン酸化していることが示唆された.
末期の拡張型心筋症のため心臓の移植をうけた患者の心筋において,Mst1の活性およびThr108のリン酸化されたBeclin1の量は,正常な心筋と比較して有意に増加していた.また,心不全を起こした心筋では正常な心筋と比較してオートファジーが抑制されており,p62が共局在するアグリソームもより多く集積していた.このように,Mst1とBeclin1との相互作用がもたらすオートファジーの抑制と変性タンパク質の集積は,心不全を起こした心筋においても生じていた.
今回,筆者らは,Mst1がBeclin1のリン酸化を介しBeclin1とBcl-2あるいはBcl-xLとの結合の強化を促進することによりVps34複合体Iの活性を抑制し,オートファジーを直接的に抑制していることを明らかにした.また,Mst1はオートファジーの抑制を介し慢性心筋梗塞を起こした心筋細胞におけるアグリソームの蓄積を促進し,心臓の機能不全を起こしていることも明らかにした.さらに,ヒトの末期拡張型心筋症の心筋においてもMst1の活性化,Beclin1のリン酸化,オートファジーの抑制,アグリソームの蓄積の増加が認められたことから,Mst1によるオートファジーの活性抑制が心不全の発症および病状の進展に関与していることが示唆された.また,Mst1によるBeclin1とBcl-2あるいはBcl-xLとの結合の強化作用は,オートファジーを抑制するだけでなく,アポトーシス促進タンパク質であるBaxのBcl-2あるいはBcl-xLからの解離によりアポトーシスを促進していることも見い出した.つまり,Mst1は心不全を起こした心臓において,オートファジーの抑制により変性タンパク質や障害をうけたオルガネラの集積を増加させる一方で,アポトーシスも促進して心臓の収縮能を低下させていると考えられた.以上より,Mst1は心不全の重要な治療標的になりうることが考えられた.
略歴:2005年 東京医科歯科大学大学院医歯学総合研究科 修了,同年 同 博士研究員,2006年 同 助教,2008年 米国Rutgers New Jersey Medical School博士研究員,2012年 同 アシスタントプロフェッサーを経て,2013年 東京医科歯科大学医学部附属病院 助教.
研究テーマ:心不全の発症および病態の進展における分子機序.
佐渡島 純一(Junichi Sadoshima)
米国Rutgers New Jersey Medical School教授.
研究室URL:http://www.sadoshimalab.org/
© 2013 前嶋康浩・佐渡島純一 Licensed under CC 表示 2.1 日本
(米国Rutgers New Jersey Medical School,Department of Cell Biology and Molecular Medicine)
email:前嶋康浩,佐渡島純一
DOI: 10.7875/first.author.2013.138
Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2.
Yasuhiro Maejima, Shiori Kyoi, Peiyong Zhai, Tong Liu, Hong Li, Andreas Ivessa, Sebastiano Sciarretta, Dominic P Del Re, Daniela K Zablocki, Chiao-Po Hsu, Dae-Sik Lim, Mitsuaki Isobe, Junichi Sadoshima
Nature Medicine, 19, 1478-1488 (2013)
要 約
筆者らは,アポトーシスを誘導するキナーゼであるMst1に,オートファジーを阻害してタンパク質の品質管理システム機構を負に制御する機能があることを見い出した.ストレスにより活性化されたMst1は心筋細胞においてオートファゴソームの形成を抑制し,p62の集積とアグリソームの蓄積を促進した.Mst1はBeclin1のBH3ドメインにあるThrをリン酸化し,Beclin1とBcl-2あるいはBcl-xLとの結合を強化してBeclin1ホモ二量体を安定化させた.その結果,Vps34複合体IのPI3キナーゼ活性が抑制されオートファゴソームの形成が阻害された.さらに,Mst1がBeclin1とBcl-2あるいはBcl-xLとの結合を促進することにより,アポトーシス促進タンパク質であるBaxのBcl-2あるいはBcl-xLからの解離が促進されアポトーシスが誘導された.心筋梗塞や拡張型心筋症における不全な心筋においてはMst1の活性が上昇し,オートファジーが抑制されアグリソームの蓄積が顕著であった.これらの結果は,Mst1がBeclin1をリン酸化してBeclin1,Bcl-2あるいはBcl-xL,Baxとの結合能を制御することにより,オートファジーとアポトーシスとを協調的に制御していることを示唆した.
はじめに
オートファジーは変性したタンパク質や障害をうけたオルガネラを除去して細胞における恒常性を維持する役割を担っている.Atg5遺伝子の欠損によりオートファジーの抑制されたマウスの心筋ではタンパク質の凝集が蓄積して心臓の機能が低下することから,オートファジーは心臓の機能の維持に欠かせないことが示唆されている1).また,ストレスにより活性化するオートファジーは心筋に対し保護的に作用することが一般的であるが2),その一方で,虚血後の再灌流のときのようにオートファジーが過剰に活性化すると心筋細胞はダメージをうける3).このように,オートファジーが生理的なレベルより抑制されること,あるいは,過剰に活性化することは,心筋細胞にとり有害である.しかし,オートファジーを最適なレベルに保つ分子機序については,いまだ解明されていない.
オートファジーの進展に重要な役割を担うタンパク質であるBeclin1は,クラスIIIのPI3キナーゼであるVps34と結合して2つの異なる複合体を形成する4).そのうち,Beclin1,Vps34,Atg14Lから構成されるVps34複合体Iは初期のオートファゴソームの形成に関与している.また,Bcl-2ファミリータンパク質であるBcl-2あるいはBcl-xLはBeclin1と結合してBeclin1-Vps34複合体の形成を阻害する5).しかし,Beclin1とBcl-2あるいはBcl-xLとの相互作用がどのような機序によりBeclin1のはたらきを抑制しているのかについては不明である.
Hippoシグナル伝達経路の主要な制御タンパク質であるセリン/スレオニンキナーゼMst1は,心筋細胞にアポトーシスが誘導されたときもっとも活性化しているキナーゼのひとつである6,7).筆者らは,これまでに,心筋においてMst1を恒常的に活性化させると拡張型心筋症が発症することや,内在性のMst1を阻害するとストレスにより誘導される心筋細胞のアポトーシスが抑制されることを見い出してきた6,8).さらに,この研究において,心筋においてMst1を恒常的に活性化させると,心筋細胞においてアグリソームやオートファジーの代表的な基質であるp62が著明に蓄積することも見い出した.
筆者らは,以上の背景より,Mst1が活性化するとオートファジーを抑制して心筋細胞に悪影響を及ぼし,心不全の発症を促すのではないかと考えた.
1.Mst1はオートファジーを阻害して心臓の機能を低下させる
筆者らは以前に,Mst1には心筋梗塞ののち左室の機能不全を進行させる作用のあることを報告した8).心不全を起こした心筋にはしばしばアグリソームの蓄積を認めるが,陳旧性心筋梗塞モデルにおける梗塞境界領域の心筋細胞にもアグリソームが著明に蓄積していた.一方,Mst1ノックアウトマウスおよび心筋に特異的にドミナントネガティブ型のMst1を過剰に発現させたトランスジェニックマウスでは,心筋梗塞ののちの心筋では,野生型マウスの心筋梗塞ののちの心筋と比べアグリソームの蓄積が著しく減少していた.また,野生型マウスの心筋梗塞ののちの心筋では,偽手術を行った心筋と比較してオートファゴソームの数が増加していたが,Mst1ノックアウトマウスの心筋梗塞ののちの心筋では,野生型マウスの心筋梗塞ののちの心筋と比較してオートファゴソームの数はさらに増加していた.これらの結果と,心筋細胞に蓄積していたアグリソームにp62が共局在していたことを考えあわせると,Mst1がオートファジーを抑制するために心筋梗塞ののちの心筋においてアグリソームが蓄積してしまうのではないかと考えた.この仮説を検証するため,Mst1ノックアウトマウスとBeclin1ヘテロノックアウトマウスとを交配させオートファジーを阻害したところ,心筋におけるアグリソームの蓄積が増加した.また,Mst1ノックアウトマウスでみられた心筋梗塞ののちの左室のリモデリングや左室の収縮能および生存率の改善の効果は,Mst1ノックアウトマウスとBeclin1ヘテロノックアウトマウスとを交配したマウスでは認められなかった.以上より,Mst1の阻害により心筋梗塞ののちに心臓の機能が改善する機序には,オートファジーの制御が関与していることが示唆された.
2.Mst1はオートファジーを抑制し細胞における変性タンパク質の蓄積を促進する
Mst1の活性化がアグリソームの形成に関与しているかどうかにつき検証した.Mst1を心筋に特異的に過剰に発現させたトランスジェニックマウスの心筋では,ポリユビキチン化タンパク質やp62が共局在するアグリソームの蓄積が増加していた.また,培養心筋細胞においてMst1はオートファジーによる長寿命タンパク質の分解を抑制した.さらに,Mst1の心筋に特異的な過剰発現トランスジェニックマウスの心筋では心筋細胞におけるオートファゴソームの数が減少していたのに対し,Mst1ノックアウトマウスではオートファゴソームの数は増加していた.以上の結果から,Mst1にはオートファジーを抑制し変性タンパク質の蓄積を促進する作用のあることが示された.オートファゴソームの蛍光のマーカーであるmRFP-GFP-LC3をアデノウイルスを用いて発現させた培養心筋細胞において,Mst1はオートリソソームの数を減少させることが確認されたことから,Mst1はオートファジーフラックスを抑制することが示唆された.
3.Mst1はVps34複合体Iの活性を直接に阻害する
Mst1によるオートファジーの制御機序につき検討した.Mst1がさまざまなオートファジー制御タンパク質と相互作用をするかどうかを免疫沈降法により検討したところ,Mst1はBeclin1と強い結合を示した.また,Mst1はBeclin1とVps34との結合,Beclin1とAtg14Lとの結合をいずれも抑制し,Vps34複合体Iのキナーゼ活性を抑制した.さらに,Mst1はBcl-2あるいはBcl-xLとBeclin1との結合を促進することも見い出した.Beclin1のBH3ドメインを模倣したABT-737は,Mst1のオートファジーの抑制作用を阻害し,Mst1によるBcl-2とBeclin1との結合の増強効果を抑制した.以上より,Mst1はBeclin1のBH3ドメインとBcl-2あるいはBcl-xLとの結合能を増強させることにより,オートファジーを抑制していることが示唆された.さらに,Mst1はBeclin1ホモ二量体の形成を促進し,Beclin1およびAtg14Lが構成するVps34複合体Iの形成を阻害していることも見い出した.
4.Mst1はBeclin1のBH3ドメインにあるThrをリン酸化しBeclin1のホモ二量体化を促進する
in vitroキナーゼアッセイにより,Mst1がBeclin1をリン酸化することが見い出された.興味深いことに,Mst1によるBeclin1のリン酸化の部位は,Bcl-2あるいはBcl-xLとの結合部位と想定されるBH3ドメインにあるThr108であることが質量分析法により判明した.実際に,内在性のMst1がBeclin1をリン酸化していることは,Thr108に特異的なリン酸化抗体を用いて確認された.また,この抗体を用いて,Mst1は小胞体に局在するBeclin1をリン酸化していることも見い出した.さらに,Mst1がBeclin1をリン酸化すると,Beclin1とBcl-2あるいはBcl-xLとの結合が強化されることも見い出した.このことは,Mst1によるリン酸化を模倣したBeclin1変異体がBcl-2あるいはBcl-xLと恒常的に強く結合すること,Mst1によりリン酸化されないBeclin1変異体ではBcl-2あるいはBcl-xLとの結合が抑制されることから確認された.Beclin1はDAPKによってもリン酸化されるが,Mst1とは逆に,DAPKはBeclin1とBcl-2あるいはBcl-xLとの解離を促進してオートファジーを誘導する.同様に,JNK1によるBcl-2あるいはBcl-xLのリン酸化もBeclin1とBcl-2あるいはBcl-xLとの解離を促進する.これらDAPKまたはJNK1によるリン酸化を介したオートファジーの活性化には,基底状態においてBeclin1とBcl-2あるいはBcl-xLとが結合している必要がある.Beclin1とBcl-2あるいはBcl-xLの結合を促進する役割を担うMst1は,Beclin1を介したオートファジーの制御に欠かせない生理的なキナーゼであると考えられた.
Mst1によるBeclin1のリン酸化がBeclin1とBcl-2あるいはBcl-xLの結合を促進する分子機構を構造生物学的な観点から検討した.Beclin1-Bcl-xL複合体の結晶構造解析によると9),Beclin1のThr108はBH3ドメインのN末端側に位置しており,Bcl-xLの形成する疎水性の溝の入口に位置するα3ヘリックスと結合する.変異タンパク質を用いたプルダウンアッセイにより,Thr108がリン酸化されて陰電荷をもつようになったBeclin1のBH3ドメインのN末端側は,Bcl-2のα3ヘリックスにある塩基性アミノ酸His117との結合が強固になり,Beclin1とBcl-xLとの結合が増強されることを見い出した.
Beclin1ホモ二量体の形成はコイルドコイルドメインを介して起こる.Beclin1ホモ二量体のコイルドコイルドメインにおける結合構造は不安定であるため,コイルドコイルドメインにおける結合構造の安定しているBeclin1-Atg14Lヘテロ二量体に遷移しやすい10).Bcl-2はBH3ドメインだけではなく,コイルドコイルドメインにおいてもBeclin1と結合すること,Beclin1のBH3ドメインとBcl-2との結合能はMst1によるリン酸化により増強されるが,Beclin1のコイルドコイルドメインはBcl-2のBH4ドメインと結合し,その結合能はMst1によるリン酸化の影響をうけないことを見い出した.このことは,Bcl-2にはMst1に依存して2つのBeclin1をつなぎとめ,Beclin1ホモ二量体を安定化させるはたらきのあることを示した.Mst1を抑制するとBeclin1とAtg14Lとの結合は強くなることから,Mst1によるBeclin1のリン酸化はBeclin1ホモ二量体を安定化させる一方で,Beclin1からのAtg14Lの解離を促進し,その結果,Vps34複合体Iの形成およびその活性化を阻害していると考えられた(図1).
5.Mst1によりリン酸化されたBeclin1はオートファジーを抑制しアポトーシスを促進する
Mst1によるBeclin1のリン酸化がオートファジーに対し及ぼす影響について検討した.Mst1によるリン酸化を模倣したBeclin1変異体を導入した心筋細胞では,Mst1の有無にかかわらずVps34複合体Iのキナーゼ活性およびオートファジーが著明に抑制された一方,Mst1によりリン酸化されないBeclin1変異体を導入した心筋細胞では,Mst1によるVps34複合体Iのキナーゼ活性の抑制は認められず,Mst1を過剰に発現させてもオートファジーの抑制はみられなかった.これらの結果より,Mst1によるBeclin1のリン酸化はオートファジーの抑制に重要なはたらきをもつことが示唆された.Mst1を心筋細胞に過剰に発現させると,Beclin1とBcl-2との結合を促進するだけでなく,Bcl-2とアポトーシス促進タンパク質であるBaxとの結合能が低下し,Baxは活性化した.Mst1によるリン酸化を模倣したBeclin1変異体を心筋細胞に導入すると,Mst1の有無にかかわらず心筋細胞のアポトーシスが促進される一方,Mst1によりリン酸化されないBeclin変異体を心筋細胞に導入すると,Mst1により誘導されるアポトーシスは抑制された.このように,Mst1により促進されるBeclin1とBcl-2との結合の増強作用は,心筋細胞においてオートファジーを抑制するだけでなく,Bcl-2からのBaxの解離および活性化を促進することにより,アポトーシスを促進するはたらきのあることが示唆された(図2).
6.心不全を起こした心筋においてBeclin1はMst1によりリン酸化されている
6週間が経過した心筋梗塞モデルマウスの心筋においてThr108のリン酸化されたBeclin1が増加していたが,Mst1ノックアウトマウスの心筋においては増加していなかったことから,心筋梗塞ののちの心筋において,内在性のMst1はBeclin1をリン酸化していることが示唆された.
末期の拡張型心筋症のため心臓の移植をうけた患者の心筋において,Mst1の活性およびThr108のリン酸化されたBeclin1の量は,正常な心筋と比較して有意に増加していた.また,心不全を起こした心筋では正常な心筋と比較してオートファジーが抑制されており,p62が共局在するアグリソームもより多く集積していた.このように,Mst1とBeclin1との相互作用がもたらすオートファジーの抑制と変性タンパク質の集積は,心不全を起こした心筋においても生じていた.
おわりに
今回,筆者らは,Mst1がBeclin1のリン酸化を介しBeclin1とBcl-2あるいはBcl-xLとの結合の強化を促進することによりVps34複合体Iの活性を抑制し,オートファジーを直接的に抑制していることを明らかにした.また,Mst1はオートファジーの抑制を介し慢性心筋梗塞を起こした心筋細胞におけるアグリソームの蓄積を促進し,心臓の機能不全を起こしていることも明らかにした.さらに,ヒトの末期拡張型心筋症の心筋においてもMst1の活性化,Beclin1のリン酸化,オートファジーの抑制,アグリソームの蓄積の増加が認められたことから,Mst1によるオートファジーの活性抑制が心不全の発症および病状の進展に関与していることが示唆された.また,Mst1によるBeclin1とBcl-2あるいはBcl-xLとの結合の強化作用は,オートファジーを抑制するだけでなく,アポトーシス促進タンパク質であるBaxのBcl-2あるいはBcl-xLからの解離によりアポトーシスを促進していることも見い出した.つまり,Mst1は心不全を起こした心臓において,オートファジーの抑制により変性タンパク質や障害をうけたオルガネラの集積を増加させる一方で,アポトーシスも促進して心臓の収縮能を低下させていると考えられた.以上より,Mst1は心不全の重要な治療標的になりうることが考えられた.
文 献
- Nakai, A., Yamaguchi, O., Takeda, T. et al.: The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med., 13, 619-624 (2007)[PubMed]
- Yan, L., Vatner, D. E., Kim, S. J. et al.: Autophagy in chronically ischemic myocardium. Proc. Natl. Acad. Sci. USA, 102, 13807-13812 (2005)[PubMed]
- Matsui, Y., Takagi, H., Qu, X. et al.: Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res., 100, 914-922 (2007)[PubMed]
- Itakura, E., Kishi, C., Inoue, K. et al.: Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell, 19, 5360-5372 (2008)[PubMed]
- Pattingre, S., Tassa, A., Qu, X. et al.: Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell, 122, 927-939 (2005)[PubMed]
- Yamamoto, S., Yang, G., Zablocki, D. et al.: Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy. J. Clin. Invest., 111, 1463-1474 (2003)[PubMed]
- Del Re, D. P., Matsuda, T., Zhai, P. et al.: Proapoptotic Rassf1A/Mst1 signaling in cardiac fibroblasts is protective against pressure overload in mice. J. Clin. Invest., 120, 3555-3567 (2010)[PubMed]
- Odashima, M., Usui, S., Takagi, H. et al.: Inhibition of endogenous Mst1 prevents apoptosis and cardiac dysfunction without affecting cardiac hypertrophy after myocardial infarction. Circ. Res., 100, 1344-1352 (2007)[PubMed]
- Oberstein, A., Jeffrey, P. D. & Shi, Y.: Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J. Biol. Chem., 282, 13123-13132 (2007)[PubMed]
- Li, X., He, L., Che, K. H., et al.: Imperfect interface of Beclin1 coiled-coil domain regulates homodimer and heterodimer formation with Atg14L and UVRAG. Nat. Commun., 3, 662 (2012)[PubMed]
著者プロフィール
略歴:2005年 東京医科歯科大学大学院医歯学総合研究科 修了,同年 同 博士研究員,2006年 同 助教,2008年 米国Rutgers New Jersey Medical School博士研究員,2012年 同 アシスタントプロフェッサーを経て,2013年 東京医科歯科大学医学部附属病院 助教.
研究テーマ:心不全の発症および病態の進展における分子機序.
佐渡島 純一(Junichi Sadoshima)
米国Rutgers New Jersey Medical School教授.
研究室URL:http://www.sadoshimalab.org/
© 2013 前嶋康浩・佐渡島純一 Licensed under CC 表示 2.1 日本
