WDR45遺伝子のde novo変異によるオートファジーの障害が神経変性症のひとつSENDAをひき起こす
村松一洋1・松本直通2
(1群馬大学大学院医学系研究科小児科学,2横浜市立大学院医学研究科遺伝学)
email:村松一洋
DOI: 10.7875/first.author.2013.029
De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood.
Hirotomo Saitsu, Taki Nishimura, Kazuhiro Muramatsu, Hirofumi Kodera, Satoko Kumada, Kenji Sugai, Emi Kasai-Yoshida, Noriko Sawaura, Hiroya Nishida, Ai Hoshino, Fukiko Ryujin, Seiichiro Yoshioka, Kiyomi Nishiyama, Yukiko Kondo, Yoshinori Tsurusaki, Mitsuko Nakashima, Noriko Miyake, Hirokazu Arakawa, Mitsuhiro Kato, Noboru Mizushima, Naomichi Matsumoto
Nature Genetics, 45, 445-449 (2013)
近年,脳に鉄の沈着をともなう神経変性症の一型で,小児期の早期からの非進行性の知的障害,成人期になり急速に進行するジストニア,パーキンソン様の症状および認知症を呈する疾患としてSENDAが提唱された.筆者らは,SENDAの2家系の全エクソーム解析からX染色体短腕に位置するWDR45遺伝子におけるde novo変異をつきとめ,SENDAの患者5名においてWDR45遺伝子の変異が原因であることを明らかにした.WDR45遺伝子はオートファジーに必須である出芽酵母Atg18のヒトにおけるホモログのひとつWIPI4をコードする.さらに,SENDAの患者のリンパ芽球においてWIPI4の発現の著しい低下,オートファジーの活性低下,および,オートファゴソームの形成障害を認めた.よって,SENDAはオートファジーにおいて中核となるタンパク質をコードする遺伝子の異常による,オートファジーの機能低下が基礎にある疾患で,ヒトにおけるオートファジーの異常と神経変性疾患との直接の関与を示すものと考えられた.
脳に鉄の沈着をともなう神経変性症は,おもに基底核へ鉄の沈着をきたし,乳児期あるいは成人期より知的障害や運動機能障害をきたすまれな進行性の神経変性疾患であり,ジストニアやパーキンソン様の症状などの錐体外路症状と知的退行を特徴とする.患者数など詳細な疫学,基底核に鉄が沈着する機序,神経変性を生じる病態や根本的な治療法など,いまだ不明な点が多い.その特徴的な経過と脳の画像の所見からいくつかの異なるタイプに分類され,パントテン酸キナーゼ関連神経変性症におけるPANK2遺伝子や神経軸索ジストロフィーにおけるPLA2G6遺伝子など,すでに原因遺伝子の同定されているタイプもあるが,依然として原因は不明で特発性として分類されるタイプも多い.このうち,小児期の早期からの非進行性の知的障害と,成人期になり急速に進行するジストニア,パーキンソン様の症状および認知症を呈し,数年で臥床状態となり,MRI画像では黒質および淡蒼球への鉄の沈着と著明な大脳の委縮を示すタイプとして,SENDA(static encephalopathy of childhood with neurodegeneration in adulthood)が知られている1-3).これまで,このSENDAには遺伝子の異常は特定されておらず,その遺伝的な原因と分子病態を明らかにし新たな予防法や治療法の開発へとつなげていくことが重要である.この研究では,SENDAの原因遺伝子の同定を目的として,SENDAの家系に対し全エクソーム解析を含む遺伝学的な解析を施行した.
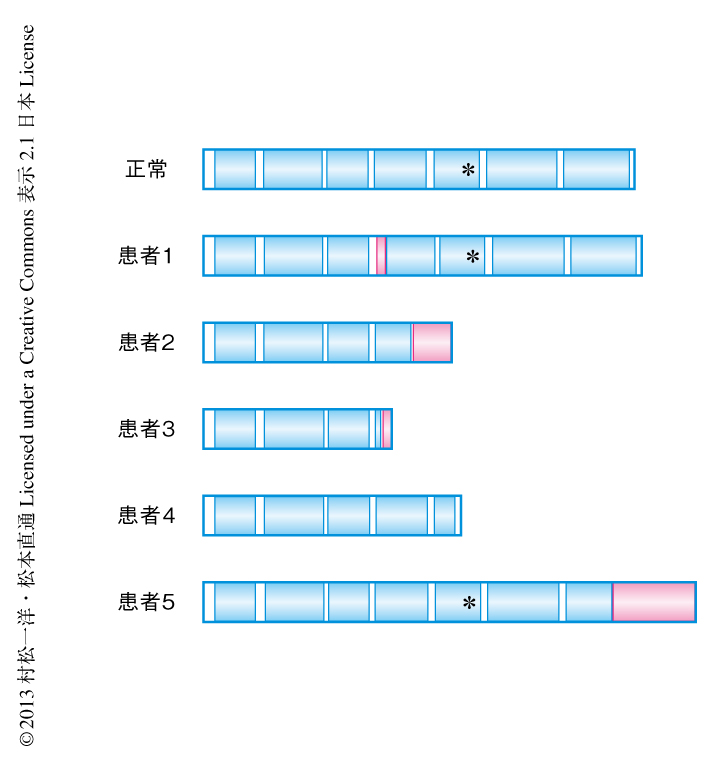
通常,SENDAは孤発性であるため優性遺伝性が疑われた.SENDAの2家系に対し全エクソーム解析を施行し原因遺伝子を探索したところ,X染色体短腕11.23に位置するWDR45遺伝子のde novo変異(両親に認めない新規の変異)を2家系で共通に見い出した.そのほか3名の患者とその家族に対しSanger法によるWDR45遺伝子の変異解析を施行したところ,すべての患者においてWDR45遺伝子の変異を認め,また,解析することのできた健常な家族にはWDR45遺伝子の変異を認めなかった.以上から,WDR45遺伝子はSENDAの原因遺伝子であると考えられた.WDR45遺伝子はオートファジーにおいて必須である出芽酵母Atg18のヒトにおけるホモログのひとつWIPI4をコードする.遺伝子変異の形式から,患者のもつ変異WIPI4は,ホスファチジルイノシトールトリスリン酸の結合配列であるFRRGモチーフを欠く短縮型,あるいは,残基付加型であることが予測された(図1).
SENDAの患者のリンパ芽球から抽出したRNAを用いてRT-PCR解析を施行したところ,4例において変異WDR45遺伝子の明らかに優位な発現を確認した.X染色体不活性化解析では4例のうち3例において著しいかたよりを認め,変異WDR45遺伝子をもたないX染色体が主として不活性化されることにより,変異WDR45遺伝子の発現が優位になっているものと考えられた(1例においては,X染色体不活性化に関する情報は得られなかった).残りの1例では正常WDR45遺伝子と変異WDR45遺伝子の両者の発現を認め,X染色体不活性化のかたよりは認めなかった.
SENDA患者のリンパ芽球から抽出したタンパク質を用いてウェスタンブロット解析を行ったところ,5名の患者すべてにおいてWIPI4の著しい発現低下を認めた.また,サイズの小さな産物など変異型と考えられるWIPI4は検出されなかった.短縮型の変異WIPI4を発現する患者では,ほかの患者に比べ重症度の高いことも想定されたが,5名の患者において明らかな重症度の差は認められなかった.したがって,いずれの変異WIPI4も分解されてしまい,WDR45遺伝子の変異の違いによる重症度の差はないことが予測された.
WDR45遺伝子のコードするWIPI4は,WIPI1~WIPI3とともに,オートファジーにおいて中核となる必須のタンパク質である出芽酵母Atg18のヒトにおけるホモログである.オートファジーは細胞における大規模な分解機構であり,不要なタンパク質などを分解することにより細胞における恒常性を維持している4).とくに,ニューロンにおいてはオートファジーが異常タンパク質の蓄積を防ぐことにより神経変性などを抑制しているものと考えられる.WIPI1~WIPI4はAtg2のホモログと結合して隔離膜(早期オートファゴソーム)の形成に重要なはたらきをもつので5),WIPI4の異常はオートファジーに影響を及ぼすことが予測された.
そこで,SENDAの患者および健常者のリンパ芽球に対しオートファジーフラックスアッセイを施行した.リソソームの機能を抑制するクロロキンの処理下では,通常はオートファゴソームがリソソームにより処理されないため,オートファゴソームのマーカーであるLC3-IIが増加する.ところが,患者のリンパ芽球ではクロロキン処理の前からすでにLC3-IIが増加しており,クロロキン処理下での増加の程度は,健常者のリンパ芽球に比べ,逆に患者のリンパ芽球では少なかった.つまり,オートファゴソームがそもそもリソソームにより処理されず蓄積していることが考えられた.さらに,オートファジーを誘導するトーリンとクロロキンの同時処理下において,健常者のリンパ芽球ではLC3-IIの増加が顕著であったが,クロロキン単独処理と同様に,患者のリンパ芽球では増加の程度は健常者のリンパ芽球に比べ少なかった.このオートファジーの誘導に対する反応が弱いということは,オートファジーの活性自体が低下していることを示すと考えられた.
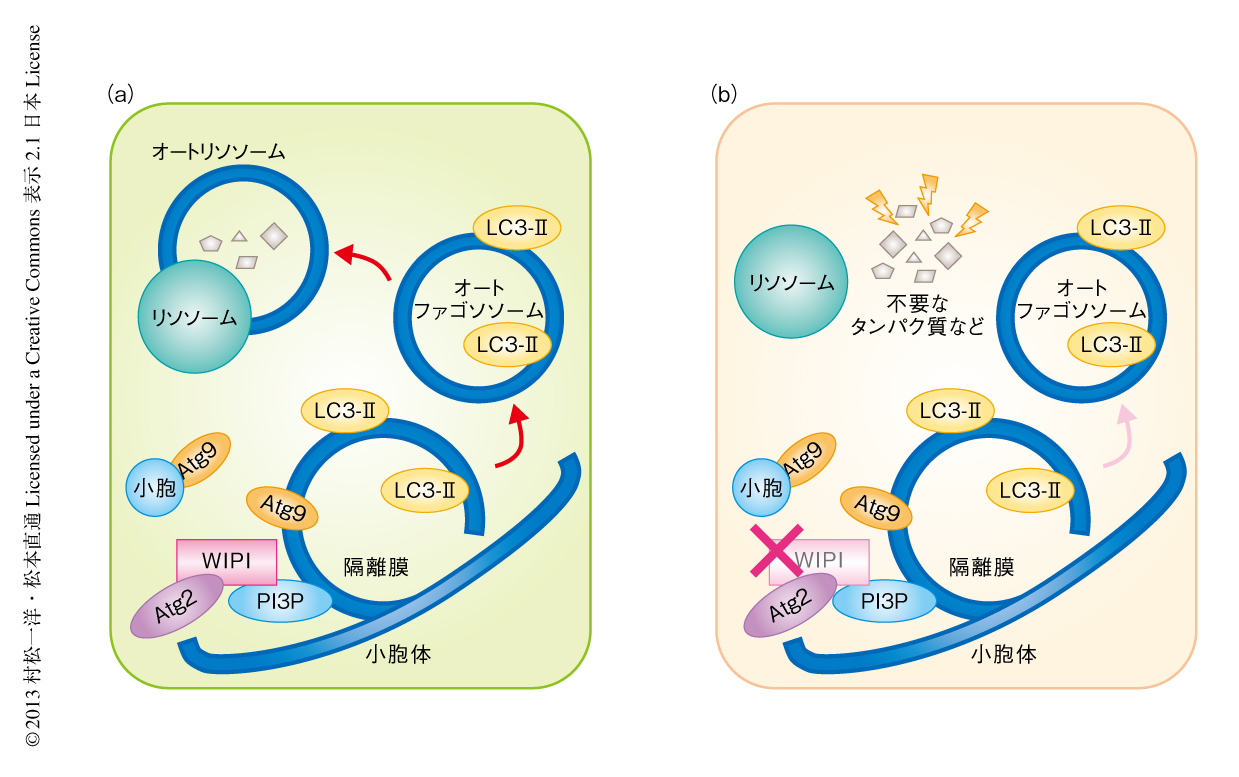
SENDAの患者のリンパ芽球での免疫蛍光染色において,オートファジーフラックスアッセイと同様にLC3-IIの蓄積が認められた.最近の研究により,ラットの腎臓に由来する細胞におけるWDR45遺伝子の機能抑制,および,線虫におけるWIPI4のホモログの変異体であるepg-6変異体では,早期オートファゴソームの蓄積することが知られている6).そして,WIPI4は早期オートファゴソーム膜への局在やその形成に重要なAtg9Aの分布を規定していると考えられている6,7).患者および健常者のリンパ芽球においてLC3-IIとAtg9Aの二重蛍光免疫染色を施行したところ,Atg9Aは後期オートファゴソームには局在しないため健常者のリンパ芽球では共局在が少なかった一方,患者のリンパ芽球ではLC3-IIとAtg9Aとが共局在する構造体が有意に多く認められた.この結果から,SENDAの患者のリンパ芽球ではオートファジーの過程が影響をうけ,多くのオートファゴソームが未完成の状態にあると考えられた.(図2)
この研究は,オートファジーにおいて中核となるタンパク質をコードするWDR45遺伝子のde novo変異がSENDAの原因であることを証明し,ヒトにおけるオートファジーの障害と神経変性疾患との直接の関連性を明らかにした点で重要である.これまで,オートファジーの機能を欠損したマウスでは神経細胞死が認められるなど8),ニューロンにおけるオートファジーの重要性は示唆されていた.また,PARK2遺伝子の変異やPINK1遺伝子の変異による家族性パーキンソン病において,オートファジーの異常が神経細胞死を導くことも示唆されていた9,10).SENDAの患者においてオートファジーの機能は完全には消失しておらず部分的に残存していた.それが,脳に鉄の沈着をともなうほかの神経変性症とは異なり,小児期には非進行性の経過をとる理由なのかもしれない.また,WDR45遺伝子は臓器に普遍的に発現しているにもかかわらずSENDAの罹患臓器は中枢神経系に顕著であることは,ニューロンの多くが分化したのち分裂しないことと関連する可能性がある.もし,リンパ芽球と同様に,X染色体不活性化のかたよりがニューロンにおいても生じているのならオートファジーの障害は明白で,分裂しないニューロンにおいて部分的に保たれていたオートファジーの機能が成人期のある時期に破たんし,そののち,症状が急速に進行し増悪するというシナリオが考えられる.
つぎの目標は,さらなる病態の解明,進行の抑制を含めた治療法への展開である.単純に考えると,オートファジーを促進させることができれば神経細胞死を回避しSENDAの発症や進行を抑制することが期待されるが,過剰なオートファジーは有害となる可能性もあり両刃の剣である.オートファジーの異常と神経変性疾患との直接的な関連を示した今回の成果により,今後,SENDAを含めたほかの神経変性疾患においてオートファジーの機能を標的とした予防法や治療の研究が展開するきっかけとなる可能性もあるだろう.
略歴:2008年 群馬大学大学院医学系研究科博士課程 修了,同 助教,ドイツPhilipps大学Marburg研究員などを経て,群馬大学大学院医学系研究科 助教.
研究テーマ:神経変性疾患における病態と小胞輸送との関連,ドーパミン神経の機能の異常と動物モデルなど.
抱負:神経疾患の病態を解明し,新たな治療法の開発への展開をめざす.
松本 直通(Naomichi Matsumoto)
横浜市立大学院医学研究科 教授.
© 2013 村松一洋・松本直通 Licensed under CC 表示 2.1 日本
(1群馬大学大学院医学系研究科小児科学,2横浜市立大学院医学研究科遺伝学)
email:村松一洋
DOI: 10.7875/first.author.2013.029
De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood.
Hirotomo Saitsu, Taki Nishimura, Kazuhiro Muramatsu, Hirofumi Kodera, Satoko Kumada, Kenji Sugai, Emi Kasai-Yoshida, Noriko Sawaura, Hiroya Nishida, Ai Hoshino, Fukiko Ryujin, Seiichiro Yoshioka, Kiyomi Nishiyama, Yukiko Kondo, Yoshinori Tsurusaki, Mitsuko Nakashima, Noriko Miyake, Hirokazu Arakawa, Mitsuhiro Kato, Noboru Mizushima, Naomichi Matsumoto
Nature Genetics, 45, 445-449 (2013)
要 約
近年,脳に鉄の沈着をともなう神経変性症の一型で,小児期の早期からの非進行性の知的障害,成人期になり急速に進行するジストニア,パーキンソン様の症状および認知症を呈する疾患としてSENDAが提唱された.筆者らは,SENDAの2家系の全エクソーム解析からX染色体短腕に位置するWDR45遺伝子におけるde novo変異をつきとめ,SENDAの患者5名においてWDR45遺伝子の変異が原因であることを明らかにした.WDR45遺伝子はオートファジーに必須である出芽酵母Atg18のヒトにおけるホモログのひとつWIPI4をコードする.さらに,SENDAの患者のリンパ芽球においてWIPI4の発現の著しい低下,オートファジーの活性低下,および,オートファゴソームの形成障害を認めた.よって,SENDAはオートファジーにおいて中核となるタンパク質をコードする遺伝子の異常による,オートファジーの機能低下が基礎にある疾患で,ヒトにおけるオートファジーの異常と神経変性疾患との直接の関与を示すものと考えられた.
はじめに
脳に鉄の沈着をともなう神経変性症は,おもに基底核へ鉄の沈着をきたし,乳児期あるいは成人期より知的障害や運動機能障害をきたすまれな進行性の神経変性疾患であり,ジストニアやパーキンソン様の症状などの錐体外路症状と知的退行を特徴とする.患者数など詳細な疫学,基底核に鉄が沈着する機序,神経変性を生じる病態や根本的な治療法など,いまだ不明な点が多い.その特徴的な経過と脳の画像の所見からいくつかの異なるタイプに分類され,パントテン酸キナーゼ関連神経変性症におけるPANK2遺伝子や神経軸索ジストロフィーにおけるPLA2G6遺伝子など,すでに原因遺伝子の同定されているタイプもあるが,依然として原因は不明で特発性として分類されるタイプも多い.このうち,小児期の早期からの非進行性の知的障害と,成人期になり急速に進行するジストニア,パーキンソン様の症状および認知症を呈し,数年で臥床状態となり,MRI画像では黒質および淡蒼球への鉄の沈着と著明な大脳の委縮を示すタイプとして,SENDA(static encephalopathy of childhood with neurodegeneration in adulthood)が知られている1-3).これまで,このSENDAには遺伝子の異常は特定されておらず,その遺伝的な原因と分子病態を明らかにし新たな予防法や治療法の開発へとつなげていくことが重要である.この研究では,SENDAの原因遺伝子の同定を目的として,SENDAの家系に対し全エクソーム解析を含む遺伝学的な解析を施行した.
1.SENDAはWDR45遺伝子のde novo変異が原因である
通常,SENDAは孤発性であるため優性遺伝性が疑われた.SENDAの2家系に対し全エクソーム解析を施行し原因遺伝子を探索したところ,X染色体短腕11.23に位置するWDR45遺伝子のde novo変異(両親に認めない新規の変異)を2家系で共通に見い出した.そのほか3名の患者とその家族に対しSanger法によるWDR45遺伝子の変異解析を施行したところ,すべての患者においてWDR45遺伝子の変異を認め,また,解析することのできた健常な家族にはWDR45遺伝子の変異を認めなかった.以上から,WDR45遺伝子はSENDAの原因遺伝子であると考えられた.WDR45遺伝子はオートファジーにおいて必須である出芽酵母Atg18のヒトにおけるホモログのひとつWIPI4をコードする.遺伝子変異の形式から,患者のもつ変異WIPI4は,ホスファチジルイノシトールトリスリン酸の結合配列であるFRRGモチーフを欠く短縮型,あるいは,残基付加型であることが予測された(図1).
2.SENDAの患者ではWIPI4の発現低下とX染色体不活性化のかたよりを認める
SENDAの患者のリンパ芽球から抽出したRNAを用いてRT-PCR解析を施行したところ,4例において変異WDR45遺伝子の明らかに優位な発現を確認した.X染色体不活性化解析では4例のうち3例において著しいかたよりを認め,変異WDR45遺伝子をもたないX染色体が主として不活性化されることにより,変異WDR45遺伝子の発現が優位になっているものと考えられた(1例においては,X染色体不活性化に関する情報は得られなかった).残りの1例では正常WDR45遺伝子と変異WDR45遺伝子の両者の発現を認め,X染色体不活性化のかたよりは認めなかった.
SENDA患者のリンパ芽球から抽出したタンパク質を用いてウェスタンブロット解析を行ったところ,5名の患者すべてにおいてWIPI4の著しい発現低下を認めた.また,サイズの小さな産物など変異型と考えられるWIPI4は検出されなかった.短縮型の変異WIPI4を発現する患者では,ほかの患者に比べ重症度の高いことも想定されたが,5名の患者において明らかな重症度の差は認められなかった.したがって,いずれの変異WIPI4も分解されてしまい,WDR45遺伝子の変異の違いによる重症度の差はないことが予測された.
3.WDR45遺伝子の変異はオートファジーを障害する
WDR45遺伝子のコードするWIPI4は,WIPI1~WIPI3とともに,オートファジーにおいて中核となる必須のタンパク質である出芽酵母Atg18のヒトにおけるホモログである.オートファジーは細胞における大規模な分解機構であり,不要なタンパク質などを分解することにより細胞における恒常性を維持している4).とくに,ニューロンにおいてはオートファジーが異常タンパク質の蓄積を防ぐことにより神経変性などを抑制しているものと考えられる.WIPI1~WIPI4はAtg2のホモログと結合して隔離膜(早期オートファゴソーム)の形成に重要なはたらきをもつので5),WIPI4の異常はオートファジーに影響を及ぼすことが予測された.
そこで,SENDAの患者および健常者のリンパ芽球に対しオートファジーフラックスアッセイを施行した.リソソームの機能を抑制するクロロキンの処理下では,通常はオートファゴソームがリソソームにより処理されないため,オートファゴソームのマーカーであるLC3-IIが増加する.ところが,患者のリンパ芽球ではクロロキン処理の前からすでにLC3-IIが増加しており,クロロキン処理下での増加の程度は,健常者のリンパ芽球に比べ,逆に患者のリンパ芽球では少なかった.つまり,オートファゴソームがそもそもリソソームにより処理されず蓄積していることが考えられた.さらに,オートファジーを誘導するトーリンとクロロキンの同時処理下において,健常者のリンパ芽球ではLC3-IIの増加が顕著であったが,クロロキン単独処理と同様に,患者のリンパ芽球では増加の程度は健常者のリンパ芽球に比べ少なかった.このオートファジーの誘導に対する反応が弱いということは,オートファジーの活性自体が低下していることを示すと考えられた.
4.WDR45遺伝子の変異はオートファジーの過程を未完の状態とする
SENDAの患者のリンパ芽球での免疫蛍光染色において,オートファジーフラックスアッセイと同様にLC3-IIの蓄積が認められた.最近の研究により,ラットの腎臓に由来する細胞におけるWDR45遺伝子の機能抑制,および,線虫におけるWIPI4のホモログの変異体であるepg-6変異体では,早期オートファゴソームの蓄積することが知られている6).そして,WIPI4は早期オートファゴソーム膜への局在やその形成に重要なAtg9Aの分布を規定していると考えられている6,7).患者および健常者のリンパ芽球においてLC3-IIとAtg9Aの二重蛍光免疫染色を施行したところ,Atg9Aは後期オートファゴソームには局在しないため健常者のリンパ芽球では共局在が少なかった一方,患者のリンパ芽球ではLC3-IIとAtg9Aとが共局在する構造体が有意に多く認められた.この結果から,SENDAの患者のリンパ芽球ではオートファジーの過程が影響をうけ,多くのオートファゴソームが未完成の状態にあると考えられた.(図2)
おわりに
この研究は,オートファジーにおいて中核となるタンパク質をコードするWDR45遺伝子のde novo変異がSENDAの原因であることを証明し,ヒトにおけるオートファジーの障害と神経変性疾患との直接の関連性を明らかにした点で重要である.これまで,オートファジーの機能を欠損したマウスでは神経細胞死が認められるなど8),ニューロンにおけるオートファジーの重要性は示唆されていた.また,PARK2遺伝子の変異やPINK1遺伝子の変異による家族性パーキンソン病において,オートファジーの異常が神経細胞死を導くことも示唆されていた9,10).SENDAの患者においてオートファジーの機能は完全には消失しておらず部分的に残存していた.それが,脳に鉄の沈着をともなうほかの神経変性症とは異なり,小児期には非進行性の経過をとる理由なのかもしれない.また,WDR45遺伝子は臓器に普遍的に発現しているにもかかわらずSENDAの罹患臓器は中枢神経系に顕著であることは,ニューロンの多くが分化したのち分裂しないことと関連する可能性がある.もし,リンパ芽球と同様に,X染色体不活性化のかたよりがニューロンにおいても生じているのならオートファジーの障害は明白で,分裂しないニューロンにおいて部分的に保たれていたオートファジーの機能が成人期のある時期に破たんし,そののち,症状が急速に進行し増悪するというシナリオが考えられる.
つぎの目標は,さらなる病態の解明,進行の抑制を含めた治療法への展開である.単純に考えると,オートファジーを促進させることができれば神経細胞死を回避しSENDAの発症や進行を抑制することが期待されるが,過剰なオートファジーは有害となる可能性もあり両刃の剣である.オートファジーの異常と神経変性疾患との直接的な関連を示した今回の成果により,今後,SENDAを含めたほかの神経変性疾患においてオートファジーの機能を標的とした予防法や治療の研究が展開するきっかけとなる可能性もあるだろう.
文 献
- Kruer, M. C., Boddaert, N., Schneider, S. A. et al.: Neuroimaging features of neurodegeneration with brain iron accumulation. AJNR Am. J. Neuroradiol., 33, 407-414 (2012)[PubMed]
- Kimura, Y., Sato, N., Sugai, K. et al.: MRI, MR spectroscopy, and diffusion tensor imaging findings in patient with static encephalopathy of childhood with neurodegeneration in adulthood (SENDA). Brain Dev., 35, 458-461 (2013)[PubMed]
- Kasai-Yoshida, E., Kumada, S., Yagishita, A. et al.: First video report of static encephalopathy of childhood with neurodegeneration in adulthood. Mov. Disord., 28, 397-399 (2013)[PubMed]
- Mizushima, N. & Komatsu, M.: Autophagy: renovation of cells and tissues. Cell, 147, 728-741 (2011)[PubMed]
- Watanabe, Y., Kobayashi, T., Yamamoto, H. et al.: Structure-based analyses reveal distinct binding sites for Atg2 and phosphoinositides in Atg18. J. Biol. Chem., 287, 31681-31690 (2012)[PubMed]
- Lu, Q., Yang, P., Huang, X. et al.: The WD40 repeat PtdIns(3)P-binding protein EPG-6 regulates progression of omegasomes to autophagosomes. Dev. Cell, 21, 343-357 (2011)[PubMed]
- Itakura, E., Kishi-Itakura, C., Koyama-Honda, I. et al.: Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J. Cell Sci., 125, 1488-1499 (2012)[PubMed]
- Hara, T., Nakamura, K., Matsui, M. et al.: Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature, 441, 885-889 (2006)[PubMed]
- Menzies, F. M., Moreau, K. & Rubinsztein, D. C.: Protein misfolding disorders and macroautophagy. Curr. Opin. Cell Biol., 23, 190-197 (2011)[PubMed]
- Kitada, T., Asakawa, S., Hattori, N. et al.: Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature, 392, 605-608 (1998)[PubMed]
著者プロフィール
略歴:2008年 群馬大学大学院医学系研究科博士課程 修了,同 助教,ドイツPhilipps大学Marburg研究員などを経て,群馬大学大学院医学系研究科 助教.
研究テーマ:神経変性疾患における病態と小胞輸送との関連,ドーパミン神経の機能の異常と動物モデルなど.
抱負:神経疾患の病態を解明し,新たな治療法の開発への展開をめざす.
松本 直通(Naomichi Matsumoto)
横浜市立大学院医学研究科 教授.
© 2013 村松一洋・松本直通 Licensed under CC 表示 2.1 日本
