RAB7L1とLRRK2は協調してニューロンにおける細胞内輸送を制御するとともにパーキンソン病の発症リスクを決定する
桑原 知樹
(米国Columbia大学Departments of Pathology and Cell Biology)
email:桑原知樹
DOI: 10.7875/first.author.2013.014
RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk.
David A. MacLeod, Herve Rhinn, Tomoki Kuwahara, Ari Zolin, Gilbert Di Paolo, Brian D. MacCabe, Karen S. Marder, Lawrence S. Honig, Lorraine N. Clark, Scott A. Small, Asa Abeliovich
Neuron, 77, 425-439 (2013)
近年,パーキンソン病を対象とした複数の全ゲノム相関解析から,いくつかの遺伝子もしくは領域が孤発性パーキンソン病の発症リスクに影響する危険因子として報告されている.しかしながら,同定された遺伝子あるいは領域が孤発性パーキンソン病の発症をもたらす分子機構,および,それらの相互関係については不明な点が多い.今回,筆者らは,パーキンソン病のリスク遺伝子の多型とトランスクリプトーム情報との関連解析を出発点として,パーキンソン病に感受性を示すLRRK2遺伝子とPARK16領域とが遺伝学的に強く相互作用してパーキンソン病の発症リスクに影響することを見い出した.両者の関係を実験的に検証するため,培養ニューロンおよびショウジョウバエを用いた解析を進め,PARK16領域に含まれる遺伝子のひとつRAB7L1遺伝子の発現により,パーキンソン病に連鎖するG2019S変異をもつLRRK2の発現による異常な表現型が回復することを発見した.一方,PARK16領域に存在するパーキンソン病のリスク多型は,RAB7L1 mRNAのスプライシングのパターンを変化させその発現量を減少させた.分子情報や既報の研究から,RAB7L1およびLRRK2は細胞内輸送経路にかかわることが想定されたが,ニューロンにおいてパーキンソン病に連鎖するG2019S変異をもつLRRK2の発現とRAB7L1の減少がともにエンドソームからゴルジ体にいたる細胞内輸送に異常をもたらし,この異常はもうひとつのまれな家族性パーキンソン病の病因遺伝子の産物である,レトロマー複合体のサブユニットのひとつVPS35の発現により回復することを明らかにした.以上より,RAB7L1とLRRK2は協調してパーキンソン病の発症リスクを制御し,その背景にある分子機構として,レトロマーを介した細胞内輸送の異常が関与することを提唱した.
パーキンソン病は加齢にともない発症する頻度の高い神経変性疾患であり,主として中脳黒質のドーパミンニューロンの変性および脱落を特徴とする.しかし,その根本的な原因はほとんど不明であり,分子レベルでの発症機構の解明が急務である.パーキンソン病の一部をしめる家族性パーキンソン病の家系の連鎖解析から,もっとも高頻度に変異の認められる責任遺伝子としてLRRK2(leucine-rich repeat kinase 2)遺伝子が同定されている1,2).一方,近年になり,孤発例のパーキンソン病患者のSNP(single nucleotide polymorphisms,1塩基多型)の情報からパーキンソン病の発症リスクに影響する遺伝子あるいは領域を探索する全ゲノム相関解析(genome-wide association study:GWAS)が進められ,LRRK2遺伝子と,もうひとつの家族性パーキンソン病の責任遺伝子でαシヌクレインをコードするSNCA遺伝子のほか,PARK16,MAPT,HLA,STK39などの複数の遺伝子あるいは領域が同定された3,4).したがって,LRRK2遺伝子やSNCA遺伝子は家族性あるいは孤発性をとわずパーキンソン病の発症に深く寄与する遺伝子と考えられるが,全ゲノム相関解析から同定されたほかの遺伝子あるいは領域がどのように孤発性パーキンソン病の発症リスクに影響するか,また,それらの相互関係や家族性パーキンソン病の責任遺伝子との関係については明らかではない.
そこで,この研究ではポスト全ゲノム相関解析のひとつの手法として,全ゲノム相関解析から同定された複数の遺伝子あるいは領域のあいだの遺伝学的な相互関係を,トランスクリプトームによる全遺伝子の発現変動の情報およびパーキンソン病の発症リスクにあたえる効果と関連させて評価することを試みた(図1).さらに,動物モデルを用いた遺伝学的な解析と培養ニューロンを用いた細胞生物学的な解析,および,ヒトの脳の試料の解析をあわせることにより,パーキンソン病の感受性遺伝子のあいだの機能的な関連を検討した.
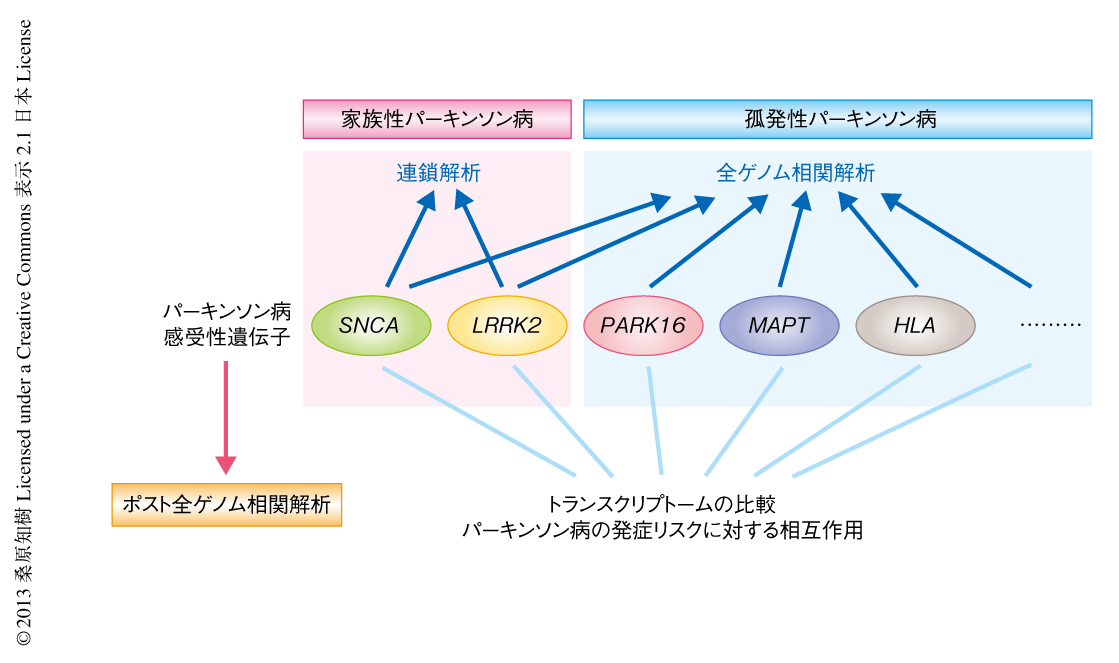
全ゲノム相関解析から同定されたパーキンソン病の感受性遺伝子が共通して表現型にあたえる影響を解析するため,パーキンソン病のリスクSNPの情報が明らかな健常人の脳における185例の遺伝子発現プロファイルを集め,それらのあいだの比較関連解析を試みた.この方法のメリットのひとつは,健常人(あるいは,未発症人)の試料のあいだで比較解析が行える点であり,パーキンソン病の発症の有無のあいだで比較する場合とは異なり,細胞死や疾患の進行にともなう2次的な影響をより抑えることができるものと考えられた.今回,おのおののSNPがトランスクリプトームにあたえる影響をGPI(global phenotypic impact)というスコアに換算して評価することにより,7つのパーキンソン病の感受性遺伝子あるいは感受性領域,LRRK2,PARK16,MAPT,SNCA,HLA-DRA,LAMP3,STK39のあいだの関連解析を行った.その結果,LRRK2遺伝子とPARK16領域がトランスクリプトームに対し共通した影響をあたえること,また,この2つに存在するSNPのGPIスコアにあたえる効果は互いに相加的ではないことを見い出した.したがって,LRRK2遺伝子とPARK16領域とが健常人の脳において遺伝学的に相互作用する可能性が示唆された.
そこで,これら2つがパーキンソン病の発症リスクに対しても遺伝学的な相互作用を示すかどうか,すなわち,1つのパーキンソン病リスクSNPがほかのパーキンソン病リスクSNPとパーキンソン病の発症リスクとの関係を制御するかどうかについて検討した.既報の4つの全ゲノム相関解析をあわせたメタ解析の結果,LRRK2遺伝子に存在するパーキンソン病リスクSNPがパーキンソン病の発症リスクにあたえる影響はPARK16領域に存在するパーキンソン病リスクSNPに強く依存しており,また,その逆も認められた.このような関係はほかのパーキンソン病のリスク遺伝子あるいはリスク領域のあいだでは弱いものであった.以上より,トランスクリプトームおよびパーキンソン病の発症リスクの両面から,LRRK2遺伝子とPARK16領域とのあいだの遺伝学的な相互作用の存在が裏づけられた.
PARK16領域には5つの遺伝子,SLC45A3遺伝子,NUCKS遺伝子,RAB7L1遺伝子,SLC41A1遺伝子,PM20D1遺伝子が存在する.そこで,どの遺伝子がLRRK2遺伝子と機能的に関連しているかを実験的に検証することを試みた.パーキンソン病に連鎖するG2019S変異をもつLRRK2の発現は,ラット初代培養ニューロンの突起を短縮させることが知られている5).5つの遺伝子の効果を検討した結果,唯一,RAB7L1遺伝子を過剰発現させた場合にG2019S変異をもつLRRK2の発現によるニューロンの突起の短縮が回復した.RAB7L1は機能未知のタンパク質であるが,その配列からRabファミリー低分子量Gタンパク質のひとつと考えられる.そこで,ほかのRabファミリータンパク質にならい恒常活性型(GTP結合型)RAB7L1変異体および不活性型(GDP結合型)RAB7L1変異体を作製したところ,恒常活性型RAB7L1変異体の発現はG2019S変異をもつLRRK2の発現によるニューロンの突起の短縮を回復させた.一方,RAB7L1のノックダウンにより,G2019S変異をもつLRRK2の発現と同様のニューロンの突起の短縮がひき起こされた.RAB7L1とLRRK2の関係をより詳細に解析したところ,免疫共沈降実験からRAB7L1とLRRK2とが細胞およびマウスの脳において結合していること,また,免疫染色実験からRAB7L1とLRRK2はおもにゴルジ体の近傍において共局在していることがわかった.
in vivoにおけるRAB7L1とLRRK2との関係をさらに調べるため,ショウジョウバエを用いた解析を行った.ドーパミン神経系にG2019S変異をもつLRRK2を発現させたショウジョウバエはドーパミンニューロンの脱落と短寿命を示す.野生型RAB7L1および恒常活性型RAB7L1変異体の発現はこれらの表現型を回復させたが,一方で,不活性型RAB7L1変異体,あるいは,ほかのRabファミリータンパク質の発現はこれらの表現型を回復させなかった.また,ショウジョウバエRAB7L1ホモログのノックダウンによってもドーパミンニューロンの脱落がひき起こされた.以上より,細胞および個体においてRAB7L1がLRRK2経路にてはたらくことが示唆された.
PARK16領域に存在するパーキンソン病リスクSNPがRAB7L1の発現にあたえる影響に注目した.ヒトの全ゲノムにわたるmRNAスプライシングの情報については,すでにリンパ芽球におけるデータが報告されている6).このデータを精査したところ,PARK16領域のパーキンソン病感受性ハプロタイプがRAB7L1 mRNAのスプライシングに影響する可能性が見い出され,また,PARK16領域に存在するSNPのひとつrs1572931がイントロン1とエキソン2との境界のスプライシング制御配列に位置していることもわかった.そこで,SNP情報の明らかなヒトの大脳皮質の試料を用いて,RAB7L1 mRNAにおけるエキソン2のスプライシングのパターンを解析した.その結果,rs1572931がパーキンソン病リスクSNPである場合にエキソン2のスキップが亢進していることがわかった.この効果が連鎖不平衡にある近傍のSNPに起因する可能性を否定するため,rs1572931の1塩基のみが異なりほかの部分は相同であるミニ遺伝子を構築し,ヒトの神経芽細胞腫SH-SY5Y細胞に導入してスプライシングのパターンを解析した.その結果,rs1572931がパーキンソン病リスクSNPの場合にやはりエキソン2のスキップの亢進を認めた.さらに,ヒトの大脳皮質の試料を用いてRAB7L1の発現量を調べた結果,健常人ではrs1572931がパーキンソン病リスクSNPである場合にRAB7L1タンパク質量は有意に減少していること,また,パーキンソン病患者の脳ではrs1572931のSNPに関係なくRAB7L1タンパク質量は減少していることを見い出した.以上より,PARK16領域に存在するパーキンソン病リスクSNPがRAB7L1の発現量を制御してパーキンソン病の発症リスクに影響していることが示された.
RAB7L1-LRRK2経路の細胞における機能についてラット初代培養ニューロンを用いて検討した.筆者らを含む複数のグループから,G2019S変異をもつLRRK2の発現はリソソームの肥大化をひき起こすことが報告されている5,7).今回,RAB7L1のノックダウンによってもリソソームの肥大化がみられること,同時に,リソソームの機能に重要な加水分解酵素の輸送を担うカチオン非依存性のマンノース6-リン酸受容体のリソソームへの局在が減少することが見い出された.一方,RAB7L1の過剰発現によりG2019S変異をもつLRRK2の発現によるリソソームの肥大とマンノース6-リン酸受容体のリソソームへの局在の低下は回復した.マンノース6-リン酸受容体はリソソームに加水分解酵素を輸送したのちエンドソームからトランスゴルジネットワークへとリサイクリングされ,その輸送はレトロマーとよばれるタンパク質複合体により制御されることが知られている8).そこで,マンノース6-リン酸受容体のゴルジ体への局在を検討したところ,G2019S変異をもつLRRK2の発現とRAB7L1のノックダウンのいずれによってもマンノース6-リン酸受容体のゴルジ体への局在は低下することがわかった.また,同様のマンノース6-リン酸受容体の局在の変化は,レトロマー複合体のサブユニットのひとつVPS35のノックダウンによってもひき起こされた(図2).
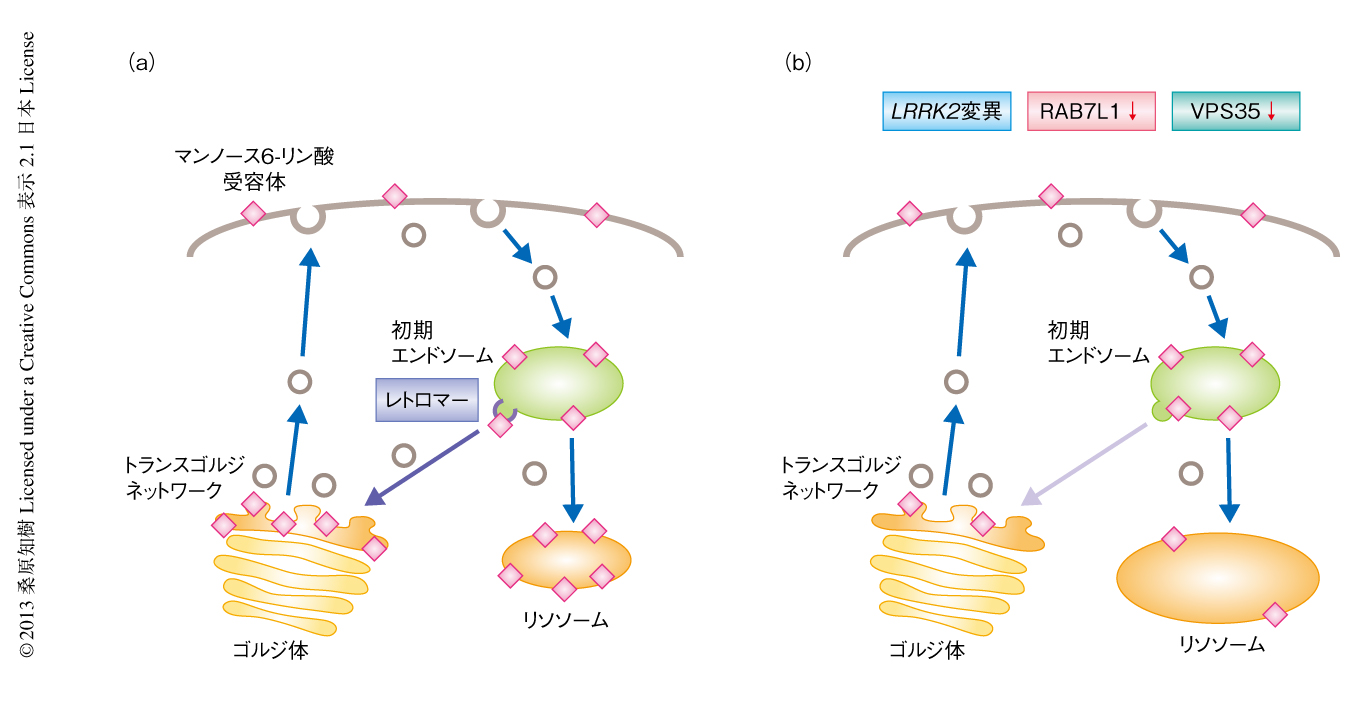
最近になり,VPS35はまれな家族性パーキンソン病の病因遺伝子の産物であることが報告されている9,10).パーキンソン病に連鎖するD620N変異をもつVPS35を過剰発現させたところ,やはり同様のマンノース6-リン酸受容体の局在の変化が認められたことから,この変異はドミナントネガティブ効果をもたらすものと考えられた.一方,野生型VPS35の過剰発現によりG2019S変異をもつLRRK2の発現によるマンノース6-リン酸受容体の局在の変化は回復した.また,野生型VPS35の過剰発現による異常な表現型の回復は,さきに述べたG2019S変異をもつLRRK2の発現やRAB7L1のノックダウンによりひき起こされるニューロンの突起の短縮や,さらには,ショウジョウバエのドーパミンニューロンの細胞死と短寿命に対しても認められた.
RAB7L1-LRRK2経路がいかにしてレトロマーの機能を制御するかについては明らかではないが,G2019S変異をもつLRRK2の発現とRAB7L1のノックダウンが共通してVPS35の発現量を減少させること,また,LRRK2とVPS35とが細胞およびマウスの脳において結合することが示された.ヒトの脳におけるVPS35の発現レベルを調べたところ,パーキンソン病患者の中脳黒質および大脳皮質においてVPS35 mRNAのレベルが有意に減少していることが見い出された.以上の結果は,パーキンソン病の分子病態にレトロマーの機能の異常が関与していることを示すものと考えられた.
これまで,パーキンソン病の発症機構に関する研究の多くが家族性パーキンソン病の遺伝子変異に着目した研究であったのに対し,筆者らは,この研究において,全ゲノム相関解析および遺伝子発現データを足がかりとして孤発性パーキンソン病の分子病態の解明を試みた.その結果,3つのパーキンソン病関連遺伝子の産物,LRRK2,RAB7L1,VPS35が同じ細胞内輸送経路において機能し,パーキンソン病の発症リスクの制御につながることを明らかにした.この3者の関係については依然として不明な点が多く残されているものの,RAB7L1あるいはレトロマーの機能亢進がパーキンソン病に連鎖するG2019S変異をもつLRRK2の発現による異常な表現型を回復させたことは,疾患研究の観点からは重要な知見であると考えられた.とくに,レトロマーを介した細胞内輸送の関与については,パーキンソン病とならび代表的な神経変性疾患であるアルツハイマー病においても示唆されている11).今後,創薬の標的経路としての可能性も含め,レトロマーにより制御される輸送経路がどの程度まで深く疾患の発症に関与するのか,さらに検討していくことが必要と考えられる.
略歴:2007年 東京大学大学院薬学系研究科にて博士号取得,田辺三菱製薬 研究員,東京大学大学院医学系研究科 博士研究員を経て,2010年より米国Columbia大学 博士研究員.
研究テーマ:パーキンソン病のモデル動物の作製と分子病態の解明.
抱負:神経変性疾患の発症にいたるプロセスを分子レベルで明らかにすることにより,いまだ確立されていない根本治療法の開発に貢献したい.
© 2013 桑原 知樹 Licensed under CC 表示 2.1 日本
(米国Columbia大学Departments of Pathology and Cell Biology)
email:桑原知樹
DOI: 10.7875/first.author.2013.014
RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk.
David A. MacLeod, Herve Rhinn, Tomoki Kuwahara, Ari Zolin, Gilbert Di Paolo, Brian D. MacCabe, Karen S. Marder, Lawrence S. Honig, Lorraine N. Clark, Scott A. Small, Asa Abeliovich
Neuron, 77, 425-439 (2013)
要 約
近年,パーキンソン病を対象とした複数の全ゲノム相関解析から,いくつかの遺伝子もしくは領域が孤発性パーキンソン病の発症リスクに影響する危険因子として報告されている.しかしながら,同定された遺伝子あるいは領域が孤発性パーキンソン病の発症をもたらす分子機構,および,それらの相互関係については不明な点が多い.今回,筆者らは,パーキンソン病のリスク遺伝子の多型とトランスクリプトーム情報との関連解析を出発点として,パーキンソン病に感受性を示すLRRK2遺伝子とPARK16領域とが遺伝学的に強く相互作用してパーキンソン病の発症リスクに影響することを見い出した.両者の関係を実験的に検証するため,培養ニューロンおよびショウジョウバエを用いた解析を進め,PARK16領域に含まれる遺伝子のひとつRAB7L1遺伝子の発現により,パーキンソン病に連鎖するG2019S変異をもつLRRK2の発現による異常な表現型が回復することを発見した.一方,PARK16領域に存在するパーキンソン病のリスク多型は,RAB7L1 mRNAのスプライシングのパターンを変化させその発現量を減少させた.分子情報や既報の研究から,RAB7L1およびLRRK2は細胞内輸送経路にかかわることが想定されたが,ニューロンにおいてパーキンソン病に連鎖するG2019S変異をもつLRRK2の発現とRAB7L1の減少がともにエンドソームからゴルジ体にいたる細胞内輸送に異常をもたらし,この異常はもうひとつのまれな家族性パーキンソン病の病因遺伝子の産物である,レトロマー複合体のサブユニットのひとつVPS35の発現により回復することを明らかにした.以上より,RAB7L1とLRRK2は協調してパーキンソン病の発症リスクを制御し,その背景にある分子機構として,レトロマーを介した細胞内輸送の異常が関与することを提唱した.
はじめに
パーキンソン病は加齢にともない発症する頻度の高い神経変性疾患であり,主として中脳黒質のドーパミンニューロンの変性および脱落を特徴とする.しかし,その根本的な原因はほとんど不明であり,分子レベルでの発症機構の解明が急務である.パーキンソン病の一部をしめる家族性パーキンソン病の家系の連鎖解析から,もっとも高頻度に変異の認められる責任遺伝子としてLRRK2(leucine-rich repeat kinase 2)遺伝子が同定されている1,2).一方,近年になり,孤発例のパーキンソン病患者のSNP(single nucleotide polymorphisms,1塩基多型)の情報からパーキンソン病の発症リスクに影響する遺伝子あるいは領域を探索する全ゲノム相関解析(genome-wide association study:GWAS)が進められ,LRRK2遺伝子と,もうひとつの家族性パーキンソン病の責任遺伝子でαシヌクレインをコードするSNCA遺伝子のほか,PARK16,MAPT,HLA,STK39などの複数の遺伝子あるいは領域が同定された3,4).したがって,LRRK2遺伝子やSNCA遺伝子は家族性あるいは孤発性をとわずパーキンソン病の発症に深く寄与する遺伝子と考えられるが,全ゲノム相関解析から同定されたほかの遺伝子あるいは領域がどのように孤発性パーキンソン病の発症リスクに影響するか,また,それらの相互関係や家族性パーキンソン病の責任遺伝子との関係については明らかではない.
そこで,この研究ではポスト全ゲノム相関解析のひとつの手法として,全ゲノム相関解析から同定された複数の遺伝子あるいは領域のあいだの遺伝学的な相互関係を,トランスクリプトームによる全遺伝子の発現変動の情報およびパーキンソン病の発症リスクにあたえる効果と関連させて評価することを試みた(図1).さらに,動物モデルを用いた遺伝学的な解析と培養ニューロンを用いた細胞生物学的な解析,および,ヒトの脳の試料の解析をあわせることにより,パーキンソン病の感受性遺伝子のあいだの機能的な関連を検討した.
1.LRRK2遺伝子とPARK16領域の遺伝子多型は遺伝学的に相互作用してパーキンソン病の発症リスクを制御する
全ゲノム相関解析から同定されたパーキンソン病の感受性遺伝子が共通して表現型にあたえる影響を解析するため,パーキンソン病のリスクSNPの情報が明らかな健常人の脳における185例の遺伝子発現プロファイルを集め,それらのあいだの比較関連解析を試みた.この方法のメリットのひとつは,健常人(あるいは,未発症人)の試料のあいだで比較解析が行える点であり,パーキンソン病の発症の有無のあいだで比較する場合とは異なり,細胞死や疾患の進行にともなう2次的な影響をより抑えることができるものと考えられた.今回,おのおののSNPがトランスクリプトームにあたえる影響をGPI(global phenotypic impact)というスコアに換算して評価することにより,7つのパーキンソン病の感受性遺伝子あるいは感受性領域,LRRK2,PARK16,MAPT,SNCA,HLA-DRA,LAMP3,STK39のあいだの関連解析を行った.その結果,LRRK2遺伝子とPARK16領域がトランスクリプトームに対し共通した影響をあたえること,また,この2つに存在するSNPのGPIスコアにあたえる効果は互いに相加的ではないことを見い出した.したがって,LRRK2遺伝子とPARK16領域とが健常人の脳において遺伝学的に相互作用する可能性が示唆された.
そこで,これら2つがパーキンソン病の発症リスクに対しても遺伝学的な相互作用を示すかどうか,すなわち,1つのパーキンソン病リスクSNPがほかのパーキンソン病リスクSNPとパーキンソン病の発症リスクとの関係を制御するかどうかについて検討した.既報の4つの全ゲノム相関解析をあわせたメタ解析の結果,LRRK2遺伝子に存在するパーキンソン病リスクSNPがパーキンソン病の発症リスクにあたえる影響はPARK16領域に存在するパーキンソン病リスクSNPに強く依存しており,また,その逆も認められた.このような関係はほかのパーキンソン病のリスク遺伝子あるいはリスク領域のあいだでは弱いものであった.以上より,トランスクリプトームおよびパーキンソン病の発症リスクの両面から,LRRK2遺伝子とPARK16領域とのあいだの遺伝学的な相互作用の存在が裏づけられた.
2.PARK16領域に存在するRAB7L1遺伝子はパーキンソン病に連鎖するG2019S変異をもつLRRK2の発現による異常な表現型を回復する
PARK16領域には5つの遺伝子,SLC45A3遺伝子,NUCKS遺伝子,RAB7L1遺伝子,SLC41A1遺伝子,PM20D1遺伝子が存在する.そこで,どの遺伝子がLRRK2遺伝子と機能的に関連しているかを実験的に検証することを試みた.パーキンソン病に連鎖するG2019S変異をもつLRRK2の発現は,ラット初代培養ニューロンの突起を短縮させることが知られている5).5つの遺伝子の効果を検討した結果,唯一,RAB7L1遺伝子を過剰発現させた場合にG2019S変異をもつLRRK2の発現によるニューロンの突起の短縮が回復した.RAB7L1は機能未知のタンパク質であるが,その配列からRabファミリー低分子量Gタンパク質のひとつと考えられる.そこで,ほかのRabファミリータンパク質にならい恒常活性型(GTP結合型)RAB7L1変異体および不活性型(GDP結合型)RAB7L1変異体を作製したところ,恒常活性型RAB7L1変異体の発現はG2019S変異をもつLRRK2の発現によるニューロンの突起の短縮を回復させた.一方,RAB7L1のノックダウンにより,G2019S変異をもつLRRK2の発現と同様のニューロンの突起の短縮がひき起こされた.RAB7L1とLRRK2の関係をより詳細に解析したところ,免疫共沈降実験からRAB7L1とLRRK2とが細胞およびマウスの脳において結合していること,また,免疫染色実験からRAB7L1とLRRK2はおもにゴルジ体の近傍において共局在していることがわかった.
in vivoにおけるRAB7L1とLRRK2との関係をさらに調べるため,ショウジョウバエを用いた解析を行った.ドーパミン神経系にG2019S変異をもつLRRK2を発現させたショウジョウバエはドーパミンニューロンの脱落と短寿命を示す.野生型RAB7L1および恒常活性型RAB7L1変異体の発現はこれらの表現型を回復させたが,一方で,不活性型RAB7L1変異体,あるいは,ほかのRabファミリータンパク質の発現はこれらの表現型を回復させなかった.また,ショウジョウバエRAB7L1ホモログのノックダウンによってもドーパミンニューロンの脱落がひき起こされた.以上より,細胞および個体においてRAB7L1がLRRK2経路にてはたらくことが示唆された.
3.PARK16領域に存在するパーキンソン病リスクSNPはRAB7L1 mRNAのスプライシングと発現を制御する
PARK16領域に存在するパーキンソン病リスクSNPがRAB7L1の発現にあたえる影響に注目した.ヒトの全ゲノムにわたるmRNAスプライシングの情報については,すでにリンパ芽球におけるデータが報告されている6).このデータを精査したところ,PARK16領域のパーキンソン病感受性ハプロタイプがRAB7L1 mRNAのスプライシングに影響する可能性が見い出され,また,PARK16領域に存在するSNPのひとつrs1572931がイントロン1とエキソン2との境界のスプライシング制御配列に位置していることもわかった.そこで,SNP情報の明らかなヒトの大脳皮質の試料を用いて,RAB7L1 mRNAにおけるエキソン2のスプライシングのパターンを解析した.その結果,rs1572931がパーキンソン病リスクSNPである場合にエキソン2のスキップが亢進していることがわかった.この効果が連鎖不平衡にある近傍のSNPに起因する可能性を否定するため,rs1572931の1塩基のみが異なりほかの部分は相同であるミニ遺伝子を構築し,ヒトの神経芽細胞腫SH-SY5Y細胞に導入してスプライシングのパターンを解析した.その結果,rs1572931がパーキンソン病リスクSNPの場合にやはりエキソン2のスキップの亢進を認めた.さらに,ヒトの大脳皮質の試料を用いてRAB7L1の発現量を調べた結果,健常人ではrs1572931がパーキンソン病リスクSNPである場合にRAB7L1タンパク質量は有意に減少していること,また,パーキンソン病患者の脳ではrs1572931のSNPに関係なくRAB7L1タンパク質量は減少していることを見い出した.以上より,PARK16領域に存在するパーキンソン病リスクSNPがRAB7L1の発現量を制御してパーキンソン病の発症リスクに影響していることが示された.
4.LRRK2とRAB7L1は協調してレトロマーを介した細胞内輸送経路を制御する
RAB7L1-LRRK2経路の細胞における機能についてラット初代培養ニューロンを用いて検討した.筆者らを含む複数のグループから,G2019S変異をもつLRRK2の発現はリソソームの肥大化をひき起こすことが報告されている5,7).今回,RAB7L1のノックダウンによってもリソソームの肥大化がみられること,同時に,リソソームの機能に重要な加水分解酵素の輸送を担うカチオン非依存性のマンノース6-リン酸受容体のリソソームへの局在が減少することが見い出された.一方,RAB7L1の過剰発現によりG2019S変異をもつLRRK2の発現によるリソソームの肥大とマンノース6-リン酸受容体のリソソームへの局在の低下は回復した.マンノース6-リン酸受容体はリソソームに加水分解酵素を輸送したのちエンドソームからトランスゴルジネットワークへとリサイクリングされ,その輸送はレトロマーとよばれるタンパク質複合体により制御されることが知られている8).そこで,マンノース6-リン酸受容体のゴルジ体への局在を検討したところ,G2019S変異をもつLRRK2の発現とRAB7L1のノックダウンのいずれによってもマンノース6-リン酸受容体のゴルジ体への局在は低下することがわかった.また,同様のマンノース6-リン酸受容体の局在の変化は,レトロマー複合体のサブユニットのひとつVPS35のノックダウンによってもひき起こされた(図2).
最近になり,VPS35はまれな家族性パーキンソン病の病因遺伝子の産物であることが報告されている9,10).パーキンソン病に連鎖するD620N変異をもつVPS35を過剰発現させたところ,やはり同様のマンノース6-リン酸受容体の局在の変化が認められたことから,この変異はドミナントネガティブ効果をもたらすものと考えられた.一方,野生型VPS35の過剰発現によりG2019S変異をもつLRRK2の発現によるマンノース6-リン酸受容体の局在の変化は回復した.また,野生型VPS35の過剰発現による異常な表現型の回復は,さきに述べたG2019S変異をもつLRRK2の発現やRAB7L1のノックダウンによりひき起こされるニューロンの突起の短縮や,さらには,ショウジョウバエのドーパミンニューロンの細胞死と短寿命に対しても認められた.
RAB7L1-LRRK2経路がいかにしてレトロマーの機能を制御するかについては明らかではないが,G2019S変異をもつLRRK2の発現とRAB7L1のノックダウンが共通してVPS35の発現量を減少させること,また,LRRK2とVPS35とが細胞およびマウスの脳において結合することが示された.ヒトの脳におけるVPS35の発現レベルを調べたところ,パーキンソン病患者の中脳黒質および大脳皮質においてVPS35 mRNAのレベルが有意に減少していることが見い出された.以上の結果は,パーキンソン病の分子病態にレトロマーの機能の異常が関与していることを示すものと考えられた.
おわりに
これまで,パーキンソン病の発症機構に関する研究の多くが家族性パーキンソン病の遺伝子変異に着目した研究であったのに対し,筆者らは,この研究において,全ゲノム相関解析および遺伝子発現データを足がかりとして孤発性パーキンソン病の分子病態の解明を試みた.その結果,3つのパーキンソン病関連遺伝子の産物,LRRK2,RAB7L1,VPS35が同じ細胞内輸送経路において機能し,パーキンソン病の発症リスクの制御につながることを明らかにした.この3者の関係については依然として不明な点が多く残されているものの,RAB7L1あるいはレトロマーの機能亢進がパーキンソン病に連鎖するG2019S変異をもつLRRK2の発現による異常な表現型を回復させたことは,疾患研究の観点からは重要な知見であると考えられた.とくに,レトロマーを介した細胞内輸送の関与については,パーキンソン病とならび代表的な神経変性疾患であるアルツハイマー病においても示唆されている11).今後,創薬の標的経路としての可能性も含め,レトロマーにより制御される輸送経路がどの程度まで深く疾患の発症に関与するのか,さらに検討していくことが必要と考えられる.
文 献
- Paisan-Ruiz, C., Jain, S., Evans, E. W. et al.: Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron, 44, 595-600 (2004)[PubMed]
- Zimprich, A., Biskup, S., Leitner, P. et al.: Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron, 44, 601-607 (2004)[PubMed]
- Satake, W., Nakabayashi, Y., Mizuta, I. et al.: Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet., 41, 1303-1307 (2009)[PubMed]
- Simon-Sanchez, J., Schulte, C., Bras, J. M. et al.: Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet., 41, 1308-1312 (2009)[PubMed]
- MacLeod, D., Dowman, J., Hammond, R. et al.: The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron, 52, 587-593 (2006)[PubMed]
- Montgomery, S. B., Sammeth, M., Gutierrez-Arcelus, M. et al.: Transcriptome genetics using second generation sequencing in a Caucasian population. Nature, 464, 773-777 (2010)[PubMed]
- Dodson, M. W., Zhang, T., Jiang, C. et al.: Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum. Mol. Genet., 21, 1350-1363 (2012)[PubMed]
- Arighi, C. N., Hartnell, L. M., Aguilar, R. C. et al.: Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor. J. Cell Biol., 165, 123-133 (2004)[PubMed]
- Vilarino-Guell, C., Wider, C., Ross, O. A. et al.: VPS35 mutations in Parkinson disease. Am. J. Hum. Genet., 89, 162-167 (2011)[PubMed]
- Zimprich, A., Benet-Pages, A., Struhal, W. et al.: A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet., 89, 168-175 (2011)[PubMed]
- Muhammad, A., Flores, I., Zhang, H. et al.: Retromer deficiency observed in Alzheimer’s disease causes hippocampal dysfunction, neurodegeneration, and Aβ accumulation. Proc. Natl. Acad. Sci. USA., 105, 7327-7332 (2008)[PubMed]
著者プロフィール
略歴:2007年 東京大学大学院薬学系研究科にて博士号取得,田辺三菱製薬 研究員,東京大学大学院医学系研究科 博士研究員を経て,2010年より米国Columbia大学 博士研究員.
研究テーマ:パーキンソン病のモデル動物の作製と分子病態の解明.
抱負:神経変性疾患の発症にいたるプロセスを分子レベルで明らかにすることにより,いまだ確立されていない根本治療法の開発に貢献したい.
© 2013 桑原 知樹 Licensed under CC 表示 2.1 日本
