セマフォリン3Aによる骨保護作用
林 幹人・中島友紀・高柳 広
(科学技術振興機構ERATO高柳オステオネットワークプロジェクト)
email:高柳 広
DOI: 10.7875/first.author.2012.057
Osteoprotection by semaphorin 3A.
Mikihito Hayashi, Tomoki Nakashima, Masahiko Taniguchi, Tatsuhiko Kodama, Atsushi Kumanogoh, Hiroshi Takayanagi
Nature, 485, 69-74 (2012)
成体において骨は,ホルモンによる制御にくわえ,さまざまな細胞間シグナル伝達因子により制御されている.破骨細胞と骨芽細胞との細胞間クロストークは骨の恒常性の維持において非常に重要だが,破骨細胞と骨芽細胞の双方の分化を制御し骨量を規定するような因子はいまだみつかっていない.筆者らは,セマフォリン3Aが破骨細胞の分化抑制と骨芽細胞の分化促進とを同時に行うことで骨保護作用を示すことを明らかにした.セマフォリン3Aはその受容体であるニューロピリン1と結合し,ITAMシグナルおよびRhoAの活性化を抑制することでRANKLにより誘導される破骨細胞の分化を抑制した.また,セマフォリン3Aは古典的Wnt経路をつうじ骨芽細胞の分化を促進し脂肪細胞の分化を抑制することも明らかになった.セマフォリン3Aノックアウトマウスおよび変異ニューロピリン1ノックインマウスのどちらも著明な骨量の低下を示した.セマフォリン3Aの投与の効果を検討したところ,破骨細胞の分化抑制および骨芽細胞の分化促進をともなう顕著な骨量の増加作用をもつことが明らかになった.以上から,セマフォリン3Aは骨関連疾患の新規の治療薬となることが期待される.
骨の恒常性はおもにカルシウム調節ホルモンなどの内分泌系により維持されていると考えられてきたが,近年の研究により,免疫系や神経系に関連した因子によっても制御されていることが明らかになってきている1,2).骨吸収と骨形成とのバランスの破綻が骨粗鬆症などの代謝性の骨疾患につながることから,このバランスを制御する分子機構の理解が新たな治療標的の確立において重要である.骨形成は骨吸収とカップリング機構によりバランスが保たれていることが知られている.現在,骨粗鬆症の治療薬として骨吸収の抑制剤が使用されているが,この場合,カップリング機構により骨形成も同時に抑制されてしまうことがありその治療効果が損なわれてしまうことから,骨吸収と骨形成の双方をバランスよく制御し骨量を回復させる薬剤および治療法の開発が望まれている.
破骨細胞は単球マクロファージ系の前駆細胞から分化する多核の巨細胞で,骨芽細胞や軟骨細胞,骨細胞などの間葉系細胞から供給されるRANKL(receptor activator of NF-κB ligand)により分化が制御されている1).骨芽細胞系の細胞ではOsteoprotegerin(Opg)という可溶性のデコイ受容体を産生することによりRANKLの作用のバランスをとっている.しかし,骨芽細胞に由来するOpg以外の破骨細胞の分化抑制因子についてはほとんど解明されていない.
マウスの頭蓋冠細胞の培養上清はRANKLおよびM-CSF(macrophage-colony stimulating factor,マクロファージコロニー刺激因子)により誘導される破骨細胞の分化を抑制する活性をもつ.Opg以外の新規の骨芽細胞の分化抑制因子の存在を確認するため,Opgを欠損した頭蓋冠細胞の培養上清の破骨細胞の分化に対する影響を検討したところ,野生型の細胞の培養上清と同じ程度ではなかったが有意な分化抑制活性をもつことを見い出した.この培養上清に含まれる分化抑制因子の同定を進めるため,陰イオン交換クロマトグラフィーにより分画を行いそれぞれの画分の破骨細胞の分化に対する影響を検討したところ,強い分化抑制活性を示す画分を発見した.この画分に含まれるタンパク質を同定するため,SDS-ポリアクリルアミドゲル電気泳動法により分離を行い,液体クロマトグラフィー-タンデム質量分析(LC-MS/MS)法によりその同定を試みた.同定されたタンパク質のうち,分泌タンパク質であり,近年,破骨細胞と骨芽細胞との相互作用にかかわることが報告されている軸索ガイダンス因子のひとつであるセマフォリン3A(semaphorin 3A:Sema3A)に着目した.
セマフォリン3Aは軸索伸長のガイダンス因子として知られるセマフォリンファミリーに属するタンパク質である3).哺乳類ではクラス3セマフォリンからクラス7セマフォリンまで約20種が同定されており,その受容体であるニューロピリン(neuropilin:Nrp)ファミリーおよびプレキシン(plexin)ファミリーと複雑なリガンド-受容体の関係で結ばれている.クラス4セマフォリンからクラス6セマフォリンは膜型タンパク質,クラス7セマフォリンはGPIアンカー型タンパク質であり,プレキシンと直接に結合するが,セマフォリン3Aを含むクラス3セマフォリンは分泌型タンパク質でありニューロピリンを介してA型プレキシンからシグナルを伝達する.セマフォリン3Aにはこれまで軸索伸長のほか免疫制御やがん細胞の増殖抑制などさまざまな機能が報告されているが,骨代謝における役割はわかっていなかった.
セマフォリン3Aを発現する組織あるいは細胞をBioGPSデータベースの検索やRT-PCR法および免疫染色法により調べたところ,全身でもとくに骨芽細胞や骨細胞において高く発現し,逆に,破骨細胞にはほとんど発現がみられなかった.また,骨芽細胞においてはセマフォリンファミリーのなかでもセマフォリン3Aがもっとも高い発現を示した.
セマフォリン3Aが骨代謝にあたえる影響を生体レベルにおいて検討するため,セマフォリン3Aノックアウトマウスの骨を解析したところ著明な骨量の低下を示し,破骨細胞の分化および骨吸収の顕著な亢進が観察された.RANKLおよびM-CSFによりin vitroにおいて骨髄マクロファージから破骨細胞の分化誘導を行なっても,野生型の細胞とセマフォリン3Aを欠損した細胞とで差はみられず,骨髄における破骨細胞前駆細胞の数にも変化はみられなかったが,セマフォリン3Aノックアウトマウスに由来する骨芽細胞様細胞を支持細胞として破骨細胞の分化を検討したところ,その著明な亢進が観察された.逆に,野生型マウスに由来する骨芽細胞様細胞を支持細胞としてセマフォリン3Aノックアウトマウスに由来する骨髄細胞を破骨細胞へと分化させても正常であったことから,骨芽細胞に由来するセマフォリン3Aが破骨細胞の分化を抑制していることが示唆された.実際に,セマフォリン3Aの組換えタンパク質の効果をRANKLおよびM-CSFによる破骨細胞の分化誘導系で検討したところ,RANKLの添加まえにセマフォリン3Aの処理を行うと用量に依存的な破骨細胞の分化抑制能を示した一方で,RANKLの添加ののちにセマフォリン3Aをくわえてもその効果はまったくみられなかった.また,骨芽細胞におけるRANKLおよびOpgのmRNAの発現や血中のタンパク質濃度はセマフォリン3Aノックアウトマウスでは変化はみられなかった.
セマフォリン3Aの受容体はニューロピリン1であるが,骨髄マクロファージにおけるニューロピリン1の発現はRANKLの刺激にともない急激かつ強力に抑制されることがわかった.このことが,RANKLの刺激ののちセマフォリン3Aの効果がみられなくなる原因となっていることが考えられた.実際に,ニューロピリン1を強制発現するとRANKLの刺激ののちにもセマフォリン3Aによる破骨細胞の分化抑制活性が確認され,逆に,ニューロピリン1をノックダウンするとその活性はまったくみられなくなった.さらに,クロマチン免疫沈降(ChIP)法やプロモーター解析などにより,ニューロピリン1の発現抑制にはRANKLにより活性化される転写因子のうち,NFATc1やc-Fosではなく,NF-κBが重要な役割をはたしていることが示された.
また,ニューロピリン1の骨代謝における役割を検討した.ニューロピリン1はVEGFシグナルにも重要でありそのノックアウトマウスは胎生致死であることから,セマフォリンとの結合領域に変異をもつニューロピリン1をノックインしたマウスを解析した.この変異ニューロピリン1ノックインマウスはセマフォリン3Aノックアウトマウスと同様に,破骨細胞の分化と骨吸収の亢進をともなった重篤な骨減少症の表現型を示した.
セマフォリン3Aにより制御される破骨細胞の分化がどのような分子機構によるのかを検討した.RANKLの刺激により誘導されるカテプシンK,酒石酸耐性酸性ホスファターゼ,NFATc1をそれぞれコードするmRNAの発現はセマフォリン3Aの処理により強く抑制された一方,RANKLの受容体であるRANKあるいはM-CSFの受容体であるc-FmsをそれぞれコードするmRNAの発現に影響はみられなかった.また,細胞増殖やアポトーシス,RANKLシグナルの下流のMAPキナーゼ活性化への影響も観察されなかった.
セマフォリン3Aの受容体複合体を形成するA型プレキシンのひとつであるプレキシンA1は,セマフォリン6Cおよびセマフォリン6Dの受容体でもあり,この場合にはニューロピリン1を必要としない.以前に,破骨細胞におけるセマフォリン6D-プレキシンA1シグナルの重要性が示されており,プレキシンA1はセマフォリン6Dをリガンドとする場合はTREM2およびDAP12と複合体を形成し,ITAMシグナルを活性化することで破骨細胞の分化を正に制御することが報告されている4).以上の事実をふまえ,セマフォリン3Aの存在のもとでは,ニューロピリン1がセマフォリン6DとのあいだでプレキシンA1を競合的に取り合うことで,結果的に破骨細胞の分化を負に制御しているのではないかという仮定をたてた.すなわち,RANKLの刺激のまえはニューロピリン1の発現が十分であることからセマフォリン3A,ニューロピリン1,プレキシンA1が複合体を形成し,TREM2およびDAP12との会合を阻害することで破骨細胞の分化を抑制するが,RANKLの刺激ののちニューロピリン1の発現が低下すると,プレキシンA1はセマフォリン6Dと結合しTREM2およびDAP12と会合できるようになるのではないかと考えた(図1a).実際に,破骨細胞前駆細胞を用いRANKLの刺激の前後で検討すると,RANKLの刺激のまえにはプレキシンA1とニューロピリン1との結合がみられたが,RANKLの刺激のあとにはその結合が低下し,逆に,TREM2とDAP12とが複合体を形成するようになった.また,セマフォリン3Aをそこにくわえると,RANKLの刺激をあたえてもプレキシンA1とニューロピリン1との結合は保たれたままであった.また,それにともない,ホスホリパーゼCγ2の活性化やCa2+シグナルの活性化にともない観察されるCa2+振動も抑制されていた.
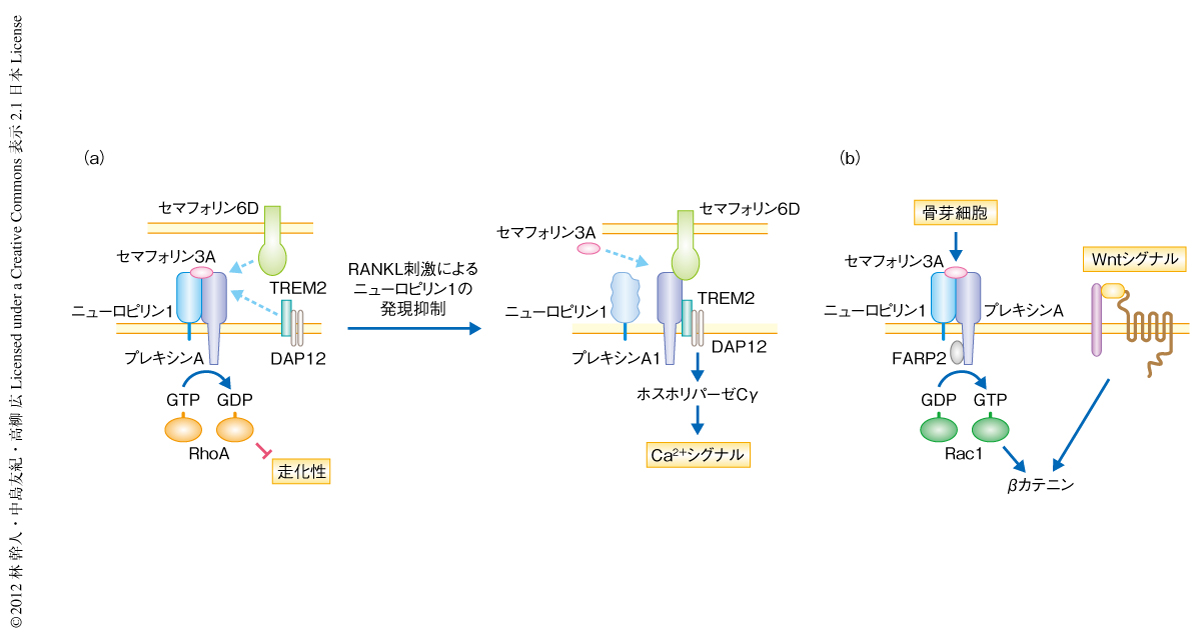
さらに,セマフォリン3AはニューロンをはじめマクロファージやT細胞などでも負の走化性を誘導することが知られていることから,破骨細胞前駆細胞の遊走能に対する影響を検討した.セマフォリン3AはM-CSFにより誘導される骨髄マクロファージの遊走を用量に依存的に抑制すること,また,この抑制は変異ニューロピリン1ノックインマウスに由来する細胞ではみられないことを確認した.セマフォリン3Aの下流ではRhoファミリーに属する低分子量Gタンパク質が制御されることが知られているが,骨髄マクロファージではM-CSFにより誘導されるRhoAの活性化の抑制によりM-CSFに対する走化性が低下することが示された(図1a).
興味深いことに,骨の解析においてセマフォリン3Aノックアウトマウスおよび変異ニューロピリン1ノックインマウスのどちらにおいても,破骨細胞の表現型にくわえ骨芽細胞と脂肪細胞にも表現型が生じることを発見した.骨芽細胞の分化や骨形成は非常に低下しており,一方で,骨髄における脂肪細胞の数の増加をみとめ,これらのマウスでは破骨細胞の分化が亢進する一方で,骨芽細胞の分化が著しく低下することで重篤な低骨量の表現型にいたっていることがわかった.また,精巣上体の脂肪組織などほかの脂肪組織における変化はみとめられなかった.
セマフォリン3Aノックアウトマウスおよび変異ニューロピリン1ノックインマウスにそれぞれ由来する頭蓋冠細胞を骨芽細胞の分化誘導培地で培養したところ,アルカリホスファターゼの活性化や骨結節の形成で確認される骨芽細胞の分化能が顕著に低下していることがわかった.また,セマフォリン3Aによる処理はセマフォリン3Aノックアウトマウスに由来する細胞における骨芽細胞への分化を促進したが,変異ニューロピリン1ノックインマウスに由来する細胞における骨芽細胞の分化に対する影響はみられなかった.一方で,セマフォリン3Aノックアウトマウスあるいは変異ニューロピリン1ノックインマウスに由来するどちらの細胞でも脂肪細胞の分化の異常な亢進がみられ,セマフォリン3Aにより処理するとセマフォリン3Aノックアウトマウスに由来する細胞においては脂肪細胞への分化が抑制されたが,この効果は変異ニューロピリン1ノックインマウスに由来する細胞ではみられなかった.
セマフォリン3Aを欠損した細胞ではRunx2やOsterix,Osteocalcinなどの骨芽細胞において重要なタンパク質をコードするmRNAの発現が著しく低下しており,逆に,PPARγやC/EBPα,aP2など脂肪細胞において重要なタンパク質のmRNAの発現は上昇していた.これらの結果から,セマフォリン3Aは受容体ニューロピリン1を介して骨芽細胞の分化を活性化し,脂肪細胞の分化を抑制することが示唆された.
セマフォリン3Aを欠損した細胞ではどのようなシグナル伝達経路が影響をうけているのかを網羅的に調べるため,セマフォリン3Aを欠損した細胞においてマイクロアレイ解析を行いgene set enrichment法により解析した.その結果,コラーゲンなどを含む細胞外マトリックスにかかわるシグナル伝達経路にくわえ,古典的Wnt経路にかかわる因子が広く影響をうけていることがわかった.古典的Wnt経路は間葉系細胞の骨芽細胞への分化を促進し脂肪細胞への分化を抑制することが既知であったことから5),この古典的Wnt経路に着目した.実際に,セマフォリン3Aを欠損した細胞ではβカテニンの転写標的として知られる因子の発現が低下していることや,古典的Wnt経路のリガンドであるWnt3aにより刺激した際に核内移行するβカテニンの量が低下していること,さらに,ここにセマフォリン3Aをくわえるとβカテニンの核内移行が促進されることが確認できた.
セマフォリン3Aシグナルの下流ではRacのグアニンヌクレオチド交換因子(guanine nucleotide exchange factor:GEF)であるFARP2というタンパク質を介しRac1が活性化されることが知られている6).Rac1は古典的Wnt経路のリガンドに応答したβカテニンの核内移行に重要であることが示されており7),この2つのシグナル伝達経路をつなぐ因子としてRac1に着目した(図1b).セマフォリン3Aを欠損した細胞におけるWnt3aの刺激ののちのRac1の活性は低下しており,セマフォリン3Aの刺激により野生型の細胞と同じ程度まで回復することが観察された.また,FARP2のドミナントネガティブ型変異体を強制発現すると,セマフォリン3Aによるβカテニンの核内移行の促進効果や骨芽細胞の分化の促進能がキャンセルされることもわかった.
セマフォリン3Aが破骨細胞の分化を抑制し骨芽細胞の分化を促進するという骨量減少性の疾患に対する治療に理想的な作用をもちあわせていることから,セマフォリン3Aの組換えタンパク質の投与の骨に対する影響を調べた.まず,5週齢の野生型マウスにセマフォリン3Aを毎週1回,4週間にわたり投与したところ,破骨細胞の分化および骨吸収の抑制,骨芽細胞の分化および骨形成の亢進をともなう骨量の有意な増加が確認された.また,この投与期間において大きな副作用などはみられなかったことから,セマフォリン3Aは骨吸収と骨形成とを同時に制御することで骨増強作用を発揮する新たな創薬標的になりうると考えられた.
さらにセマフォリン3Aの治療効果を検討するため,皮質骨にドリルにより穴を開けそこでの骨再生を調べるモデルと,卵巣の摘出による骨粗鬆症モデルに対して,セマフォリン3Aを投与した.どちらの場合でも,セマフォリン3Aは破骨細胞の分化抑制および骨芽細胞の分化促進を同時に実現することにより顕著な骨量の増加作用をもつことが明らかになった.以上の結果から,セマフォリン3Aは骨吸収を抑制すると同時に骨形成を促進することで,カップリング機構の影響をうけにくい骨関連疾患の治療薬となりうることが期待された.
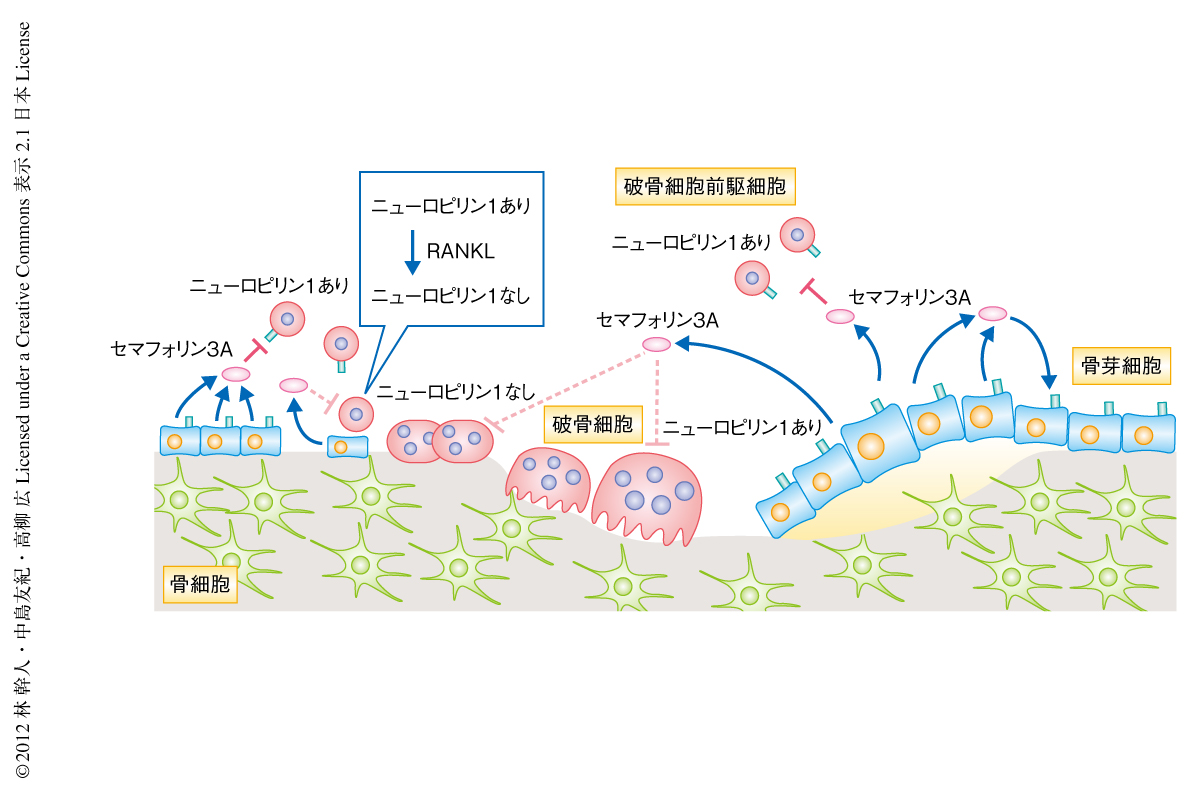
今回の報告により,骨芽細胞で発現するセマフォリン3Aは骨芽細胞と破骨細胞とを同時に制御することで骨量を増加させるはたらきをもつことが明らかになった(図2).セマフォリン3Aは骨リモデリングの骨形成期において,骨芽細胞が骨基質を産生し,破骨細胞が骨形成の場に近づかないようにするため必要な因子であることが示唆された(図2).セマフォリン3Aの血中における濃度は老齢なマウスでかなり低下していたことから,バイオマーカーとなりうる可能性もある.
骨量を増加させることのできる薬剤の候補は,現在のところ,副甲状腺ホルモン製剤と抗スクレロスチン抗体などであるが,いずれも骨芽細胞が標的であり破骨細胞に対する作用はよくわかっていない.セマフォリン3Aは骨吸収抑制薬と骨形成促進薬の2つの側面を兼ね備えた理想的な薬剤として,今後の治療応用へむけた研究が望まれる.
略歴:2010年 東京医科歯科大学大学院医歯学総合研究科 修了,同年より科学技術振興機構ERATO高柳オステオネットワークプロジェクト 研究員.
研究テーマ:骨生物学,骨免疫学.
関心事:骨と他臓器のクロストーク.
中島 友紀(Tomoki Nakashima)
東京医科歯科大学大学院医歯学総合研究科 助教.科学技術振興機構ERATO高柳オステオネットワークプロジェクト グループリーダー 兼任.
高柳 広(Hiroshi Takayanagi)
東京大学大学院医学系研究科 教授.科学技術振興機構ERATO高柳オステオネットワークプロジェクト 研究総括 兼任.
研究室URL:http://osteoimmunology.com/index.html
© 2012 林 幹人・中島友紀・高柳 広 Licensed under CC 表示 2.1 日本
(科学技術振興機構ERATO高柳オステオネットワークプロジェクト)
email:高柳 広
DOI: 10.7875/first.author.2012.057
Osteoprotection by semaphorin 3A.
Mikihito Hayashi, Tomoki Nakashima, Masahiko Taniguchi, Tatsuhiko Kodama, Atsushi Kumanogoh, Hiroshi Takayanagi
Nature, 485, 69-74 (2012)
要 約
成体において骨は,ホルモンによる制御にくわえ,さまざまな細胞間シグナル伝達因子により制御されている.破骨細胞と骨芽細胞との細胞間クロストークは骨の恒常性の維持において非常に重要だが,破骨細胞と骨芽細胞の双方の分化を制御し骨量を規定するような因子はいまだみつかっていない.筆者らは,セマフォリン3Aが破骨細胞の分化抑制と骨芽細胞の分化促進とを同時に行うことで骨保護作用を示すことを明らかにした.セマフォリン3Aはその受容体であるニューロピリン1と結合し,ITAMシグナルおよびRhoAの活性化を抑制することでRANKLにより誘導される破骨細胞の分化を抑制した.また,セマフォリン3Aは古典的Wnt経路をつうじ骨芽細胞の分化を促進し脂肪細胞の分化を抑制することも明らかになった.セマフォリン3Aノックアウトマウスおよび変異ニューロピリン1ノックインマウスのどちらも著明な骨量の低下を示した.セマフォリン3Aの投与の効果を検討したところ,破骨細胞の分化抑制および骨芽細胞の分化促進をともなう顕著な骨量の増加作用をもつことが明らかになった.以上から,セマフォリン3Aは骨関連疾患の新規の治療薬となることが期待される.
はじめに
骨の恒常性はおもにカルシウム調節ホルモンなどの内分泌系により維持されていると考えられてきたが,近年の研究により,免疫系や神経系に関連した因子によっても制御されていることが明らかになってきている1,2).骨吸収と骨形成とのバランスの破綻が骨粗鬆症などの代謝性の骨疾患につながることから,このバランスを制御する分子機構の理解が新たな治療標的の確立において重要である.骨形成は骨吸収とカップリング機構によりバランスが保たれていることが知られている.現在,骨粗鬆症の治療薬として骨吸収の抑制剤が使用されているが,この場合,カップリング機構により骨形成も同時に抑制されてしまうことがありその治療効果が損なわれてしまうことから,骨吸収と骨形成の双方をバランスよく制御し骨量を回復させる薬剤および治療法の開発が望まれている.
破骨細胞は単球マクロファージ系の前駆細胞から分化する多核の巨細胞で,骨芽細胞や軟骨細胞,骨細胞などの間葉系細胞から供給されるRANKL(receptor activator of NF-κB ligand)により分化が制御されている1).骨芽細胞系の細胞ではOsteoprotegerin(Opg)という可溶性のデコイ受容体を産生することによりRANKLの作用のバランスをとっている.しかし,骨芽細胞に由来するOpg以外の破骨細胞の分化抑制因子についてはほとんど解明されていない.
1.セマフォリン3Aは骨芽細胞に由来する破骨細胞の分化抑制因子である
マウスの頭蓋冠細胞の培養上清はRANKLおよびM-CSF(macrophage-colony stimulating factor,マクロファージコロニー刺激因子)により誘導される破骨細胞の分化を抑制する活性をもつ.Opg以外の新規の骨芽細胞の分化抑制因子の存在を確認するため,Opgを欠損した頭蓋冠細胞の培養上清の破骨細胞の分化に対する影響を検討したところ,野生型の細胞の培養上清と同じ程度ではなかったが有意な分化抑制活性をもつことを見い出した.この培養上清に含まれる分化抑制因子の同定を進めるため,陰イオン交換クロマトグラフィーにより分画を行いそれぞれの画分の破骨細胞の分化に対する影響を検討したところ,強い分化抑制活性を示す画分を発見した.この画分に含まれるタンパク質を同定するため,SDS-ポリアクリルアミドゲル電気泳動法により分離を行い,液体クロマトグラフィー-タンデム質量分析(LC-MS/MS)法によりその同定を試みた.同定されたタンパク質のうち,分泌タンパク質であり,近年,破骨細胞と骨芽細胞との相互作用にかかわることが報告されている軸索ガイダンス因子のひとつであるセマフォリン3A(semaphorin 3A:Sema3A)に着目した.
セマフォリン3Aは軸索伸長のガイダンス因子として知られるセマフォリンファミリーに属するタンパク質である3).哺乳類ではクラス3セマフォリンからクラス7セマフォリンまで約20種が同定されており,その受容体であるニューロピリン(neuropilin:Nrp)ファミリーおよびプレキシン(plexin)ファミリーと複雑なリガンド-受容体の関係で結ばれている.クラス4セマフォリンからクラス6セマフォリンは膜型タンパク質,クラス7セマフォリンはGPIアンカー型タンパク質であり,プレキシンと直接に結合するが,セマフォリン3Aを含むクラス3セマフォリンは分泌型タンパク質でありニューロピリンを介してA型プレキシンからシグナルを伝達する.セマフォリン3Aにはこれまで軸索伸長のほか免疫制御やがん細胞の増殖抑制などさまざまな機能が報告されているが,骨代謝における役割はわかっていなかった.
セマフォリン3Aを発現する組織あるいは細胞をBioGPSデータベースの検索やRT-PCR法および免疫染色法により調べたところ,全身でもとくに骨芽細胞や骨細胞において高く発現し,逆に,破骨細胞にはほとんど発現がみられなかった.また,骨芽細胞においてはセマフォリンファミリーのなかでもセマフォリン3Aがもっとも高い発現を示した.
セマフォリン3Aが骨代謝にあたえる影響を生体レベルにおいて検討するため,セマフォリン3Aノックアウトマウスの骨を解析したところ著明な骨量の低下を示し,破骨細胞の分化および骨吸収の顕著な亢進が観察された.RANKLおよびM-CSFによりin vitroにおいて骨髄マクロファージから破骨細胞の分化誘導を行なっても,野生型の細胞とセマフォリン3Aを欠損した細胞とで差はみられず,骨髄における破骨細胞前駆細胞の数にも変化はみられなかったが,セマフォリン3Aノックアウトマウスに由来する骨芽細胞様細胞を支持細胞として破骨細胞の分化を検討したところ,その著明な亢進が観察された.逆に,野生型マウスに由来する骨芽細胞様細胞を支持細胞としてセマフォリン3Aノックアウトマウスに由来する骨髄細胞を破骨細胞へと分化させても正常であったことから,骨芽細胞に由来するセマフォリン3Aが破骨細胞の分化を抑制していることが示唆された.実際に,セマフォリン3Aの組換えタンパク質の効果をRANKLおよびM-CSFによる破骨細胞の分化誘導系で検討したところ,RANKLの添加まえにセマフォリン3Aの処理を行うと用量に依存的な破骨細胞の分化抑制能を示した一方で,RANKLの添加ののちにセマフォリン3Aをくわえてもその効果はまったくみられなかった.また,骨芽細胞におけるRANKLおよびOpgのmRNAの発現や血中のタンパク質濃度はセマフォリン3Aノックアウトマウスでは変化はみられなかった.
セマフォリン3Aの受容体はニューロピリン1であるが,骨髄マクロファージにおけるニューロピリン1の発現はRANKLの刺激にともない急激かつ強力に抑制されることがわかった.このことが,RANKLの刺激ののちセマフォリン3Aの効果がみられなくなる原因となっていることが考えられた.実際に,ニューロピリン1を強制発現するとRANKLの刺激ののちにもセマフォリン3Aによる破骨細胞の分化抑制活性が確認され,逆に,ニューロピリン1をノックダウンするとその活性はまったくみられなくなった.さらに,クロマチン免疫沈降(ChIP)法やプロモーター解析などにより,ニューロピリン1の発現抑制にはRANKLにより活性化される転写因子のうち,NFATc1やc-Fosではなく,NF-κBが重要な役割をはたしていることが示された.
また,ニューロピリン1の骨代謝における役割を検討した.ニューロピリン1はVEGFシグナルにも重要でありそのノックアウトマウスは胎生致死であることから,セマフォリンとの結合領域に変異をもつニューロピリン1をノックインしたマウスを解析した.この変異ニューロピリン1ノックインマウスはセマフォリン3Aノックアウトマウスと同様に,破骨細胞の分化と骨吸収の亢進をともなった重篤な骨減少症の表現型を示した.
2.セマフォリン3Aによる破骨細胞の分化抑制の分子機構
セマフォリン3Aにより制御される破骨細胞の分化がどのような分子機構によるのかを検討した.RANKLの刺激により誘導されるカテプシンK,酒石酸耐性酸性ホスファターゼ,NFATc1をそれぞれコードするmRNAの発現はセマフォリン3Aの処理により強く抑制された一方,RANKLの受容体であるRANKあるいはM-CSFの受容体であるc-FmsをそれぞれコードするmRNAの発現に影響はみられなかった.また,細胞増殖やアポトーシス,RANKLシグナルの下流のMAPキナーゼ活性化への影響も観察されなかった.
セマフォリン3Aの受容体複合体を形成するA型プレキシンのひとつであるプレキシンA1は,セマフォリン6Cおよびセマフォリン6Dの受容体でもあり,この場合にはニューロピリン1を必要としない.以前に,破骨細胞におけるセマフォリン6D-プレキシンA1シグナルの重要性が示されており,プレキシンA1はセマフォリン6Dをリガンドとする場合はTREM2およびDAP12と複合体を形成し,ITAMシグナルを活性化することで破骨細胞の分化を正に制御することが報告されている4).以上の事実をふまえ,セマフォリン3Aの存在のもとでは,ニューロピリン1がセマフォリン6DとのあいだでプレキシンA1を競合的に取り合うことで,結果的に破骨細胞の分化を負に制御しているのではないかという仮定をたてた.すなわち,RANKLの刺激のまえはニューロピリン1の発現が十分であることからセマフォリン3A,ニューロピリン1,プレキシンA1が複合体を形成し,TREM2およびDAP12との会合を阻害することで破骨細胞の分化を抑制するが,RANKLの刺激ののちニューロピリン1の発現が低下すると,プレキシンA1はセマフォリン6Dと結合しTREM2およびDAP12と会合できるようになるのではないかと考えた(図1a).実際に,破骨細胞前駆細胞を用いRANKLの刺激の前後で検討すると,RANKLの刺激のまえにはプレキシンA1とニューロピリン1との結合がみられたが,RANKLの刺激のあとにはその結合が低下し,逆に,TREM2とDAP12とが複合体を形成するようになった.また,セマフォリン3Aをそこにくわえると,RANKLの刺激をあたえてもプレキシンA1とニューロピリン1との結合は保たれたままであった.また,それにともない,ホスホリパーゼCγ2の活性化やCa2+シグナルの活性化にともない観察されるCa2+振動も抑制されていた.
さらに,セマフォリン3AはニューロンをはじめマクロファージやT細胞などでも負の走化性を誘導することが知られていることから,破骨細胞前駆細胞の遊走能に対する影響を検討した.セマフォリン3AはM-CSFにより誘導される骨髄マクロファージの遊走を用量に依存的に抑制すること,また,この抑制は変異ニューロピリン1ノックインマウスに由来する細胞ではみられないことを確認した.セマフォリン3Aの下流ではRhoファミリーに属する低分子量Gタンパク質が制御されることが知られているが,骨髄マクロファージではM-CSFにより誘導されるRhoAの活性化の抑制によりM-CSFに対する走化性が低下することが示された(図1a).
3.セマフォリン3Aは古典的Wnt経路を介し骨芽細胞の分化を制御する
興味深いことに,骨の解析においてセマフォリン3Aノックアウトマウスおよび変異ニューロピリン1ノックインマウスのどちらにおいても,破骨細胞の表現型にくわえ骨芽細胞と脂肪細胞にも表現型が生じることを発見した.骨芽細胞の分化や骨形成は非常に低下しており,一方で,骨髄における脂肪細胞の数の増加をみとめ,これらのマウスでは破骨細胞の分化が亢進する一方で,骨芽細胞の分化が著しく低下することで重篤な低骨量の表現型にいたっていることがわかった.また,精巣上体の脂肪組織などほかの脂肪組織における変化はみとめられなかった.
セマフォリン3Aノックアウトマウスおよび変異ニューロピリン1ノックインマウスにそれぞれ由来する頭蓋冠細胞を骨芽細胞の分化誘導培地で培養したところ,アルカリホスファターゼの活性化や骨結節の形成で確認される骨芽細胞の分化能が顕著に低下していることがわかった.また,セマフォリン3Aによる処理はセマフォリン3Aノックアウトマウスに由来する細胞における骨芽細胞への分化を促進したが,変異ニューロピリン1ノックインマウスに由来する細胞における骨芽細胞の分化に対する影響はみられなかった.一方で,セマフォリン3Aノックアウトマウスあるいは変異ニューロピリン1ノックインマウスに由来するどちらの細胞でも脂肪細胞の分化の異常な亢進がみられ,セマフォリン3Aにより処理するとセマフォリン3Aノックアウトマウスに由来する細胞においては脂肪細胞への分化が抑制されたが,この効果は変異ニューロピリン1ノックインマウスに由来する細胞ではみられなかった.
セマフォリン3Aを欠損した細胞ではRunx2やOsterix,Osteocalcinなどの骨芽細胞において重要なタンパク質をコードするmRNAの発現が著しく低下しており,逆に,PPARγやC/EBPα,aP2など脂肪細胞において重要なタンパク質のmRNAの発現は上昇していた.これらの結果から,セマフォリン3Aは受容体ニューロピリン1を介して骨芽細胞の分化を活性化し,脂肪細胞の分化を抑制することが示唆された.
セマフォリン3Aを欠損した細胞ではどのようなシグナル伝達経路が影響をうけているのかを網羅的に調べるため,セマフォリン3Aを欠損した細胞においてマイクロアレイ解析を行いgene set enrichment法により解析した.その結果,コラーゲンなどを含む細胞外マトリックスにかかわるシグナル伝達経路にくわえ,古典的Wnt経路にかかわる因子が広く影響をうけていることがわかった.古典的Wnt経路は間葉系細胞の骨芽細胞への分化を促進し脂肪細胞への分化を抑制することが既知であったことから5),この古典的Wnt経路に着目した.実際に,セマフォリン3Aを欠損した細胞ではβカテニンの転写標的として知られる因子の発現が低下していることや,古典的Wnt経路のリガンドであるWnt3aにより刺激した際に核内移行するβカテニンの量が低下していること,さらに,ここにセマフォリン3Aをくわえるとβカテニンの核内移行が促進されることが確認できた.
セマフォリン3Aシグナルの下流ではRacのグアニンヌクレオチド交換因子(guanine nucleotide exchange factor:GEF)であるFARP2というタンパク質を介しRac1が活性化されることが知られている6).Rac1は古典的Wnt経路のリガンドに応答したβカテニンの核内移行に重要であることが示されており7),この2つのシグナル伝達経路をつなぐ因子としてRac1に着目した(図1b).セマフォリン3Aを欠損した細胞におけるWnt3aの刺激ののちのRac1の活性は低下しており,セマフォリン3Aの刺激により野生型の細胞と同じ程度まで回復することが観察された.また,FARP2のドミナントネガティブ型変異体を強制発現すると,セマフォリン3Aによるβカテニンの核内移行の促進効果や骨芽細胞の分化の促進能がキャンセルされることもわかった.
4.セマフォリン3Aによる骨保護作用
セマフォリン3Aが破骨細胞の分化を抑制し骨芽細胞の分化を促進するという骨量減少性の疾患に対する治療に理想的な作用をもちあわせていることから,セマフォリン3Aの組換えタンパク質の投与の骨に対する影響を調べた.まず,5週齢の野生型マウスにセマフォリン3Aを毎週1回,4週間にわたり投与したところ,破骨細胞の分化および骨吸収の抑制,骨芽細胞の分化および骨形成の亢進をともなう骨量の有意な増加が確認された.また,この投与期間において大きな副作用などはみられなかったことから,セマフォリン3Aは骨吸収と骨形成とを同時に制御することで骨増強作用を発揮する新たな創薬標的になりうると考えられた.
さらにセマフォリン3Aの治療効果を検討するため,皮質骨にドリルにより穴を開けそこでの骨再生を調べるモデルと,卵巣の摘出による骨粗鬆症モデルに対して,セマフォリン3Aを投与した.どちらの場合でも,セマフォリン3Aは破骨細胞の分化抑制および骨芽細胞の分化促進を同時に実現することにより顕著な骨量の増加作用をもつことが明らかになった.以上の結果から,セマフォリン3Aは骨吸収を抑制すると同時に骨形成を促進することで,カップリング機構の影響をうけにくい骨関連疾患の治療薬となりうることが期待された.
おわりに
今回の報告により,骨芽細胞で発現するセマフォリン3Aは骨芽細胞と破骨細胞とを同時に制御することで骨量を増加させるはたらきをもつことが明らかになった(図2).セマフォリン3Aは骨リモデリングの骨形成期において,骨芽細胞が骨基質を産生し,破骨細胞が骨形成の場に近づかないようにするため必要な因子であることが示唆された(図2).セマフォリン3Aの血中における濃度は老齢なマウスでかなり低下していたことから,バイオマーカーとなりうる可能性もある.
骨量を増加させることのできる薬剤の候補は,現在のところ,副甲状腺ホルモン製剤と抗スクレロスチン抗体などであるが,いずれも骨芽細胞が標的であり破骨細胞に対する作用はよくわかっていない.セマフォリン3Aは骨吸収抑制薬と骨形成促進薬の2つの側面を兼ね備えた理想的な薬剤として,今後の治療応用へむけた研究が望まれる.
文 献
- Takayanagi, H.: Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat. Rev. Immunol., 7, 292-304 (2007)[PubMed]
- Elefteriou, F.: Regulation of bone remodeling by the central and peripheral nervous system. Arch. Biochem. Biophys., 473, 231-236 (2008)[PubMed]
- Neufeld, G. & Kessler, O.: The semaphorins: versatile regulators of tumour progression and tumour angiogenesis. Nat. Rev. Cancer, 8, 632-645 (2008)[PubMed]
- Takegahara, N., Takamatsu, H., Toyofuku, T. et al.: Plexin-A1 and its interaction with DAP12 in immune responses and bone homeostasis. Nat. Cell Biol., 8, 615-622 (2006)[PubMed]
- Krishnan, V., Bryant, H. U. & Macdougald, O. A.: Regulation of bone mass by Wnt signaling. J. Clin. Invest., 116, 1202-1209 (2006)[PubMed]
- Toyofuku, T., Yoshida, J., Sugimoto, T. et al.: FARP2 triggers signals for Sema3A-mediated axonal repulsion. Nat. Neurosci., 8, 1712-1719 (2005)[PubMed]
- Wu, X., Tu, X., Joeng, K. S. et al.: Rac1 activation controls nuclear localization of β-catenin during canonical Wnt signaling. Cell, 133, 340-353 (2008)[PubMed]
著者プロフィール
略歴:2010年 東京医科歯科大学大学院医歯学総合研究科 修了,同年より科学技術振興機構ERATO高柳オステオネットワークプロジェクト 研究員.
研究テーマ:骨生物学,骨免疫学.
関心事:骨と他臓器のクロストーク.
中島 友紀(Tomoki Nakashima)
東京医科歯科大学大学院医歯学総合研究科 助教.科学技術振興機構ERATO高柳オステオネットワークプロジェクト グループリーダー 兼任.
高柳 広(Hiroshi Takayanagi)
東京大学大学院医学系研究科 教授.科学技術振興機構ERATO高柳オステオネットワークプロジェクト 研究総括 兼任.
研究室URL:http://osteoimmunology.com/index.html
© 2012 林 幹人・中島友紀・高柳 広 Licensed under CC 表示 2.1 日本
