ユビキチンリガーゼZNRF1はAKTをプロテアソーム依存的に分解しGSK3BによるCRMP2のリン酸化を誘導することにより神経軸索の変性を促進する
若月修二・荒木敏之
(国立精神・神経医療研究センター神経研究所 疾病研究第5部)
email:若月修二,荒木敏之
DOI: 10.7875/first.author.2011.177
ZNRF1 promotes Wallerian degeneration by degrading AKT to induce GSK3B-dependent CRMP2 phosphorylation.
Shuji Wakatsuki, Fuminori Saitoh, Toshiyuki Araki
Nature Cell Biology, 13, 1415-1423 (2011)
神経軸索の変性は多くの神経変性疾患に共通する所見であり,その制御機構を明らかにすることは疾患の抑制,さらには治療方法の開発に重要な意味をもつものと考えられる.今回,筆者らは,セリン-スレオニンキナーゼであるAKTがユビキチンリガーゼZNRF1を介しプロテアソームに依存的に分解されて軸索から消失することが,軸索の変性を促進することを明らかにした.AKTにはリン酸化を介しGSK3Bを抑制するはたらきがあり,軸索の変性が開始されるとAKTの分解によりこの抑制が失われ,活性化したGSK3Bがリン酸化を介しCRMP2を不活性化することが微小管の崩壊を促進し軸索の変性を進行させるものと考えられた.これらの結果は,AKTを介したリン酸化シグナル伝達カスケードが軸索の安定性や変性を制御する主要な反応経路であることを示すとともに,この反応経路を抑制する方法を開発することが神経疾患の症状改善や進行の抑制につながる可能性を強く示唆した.
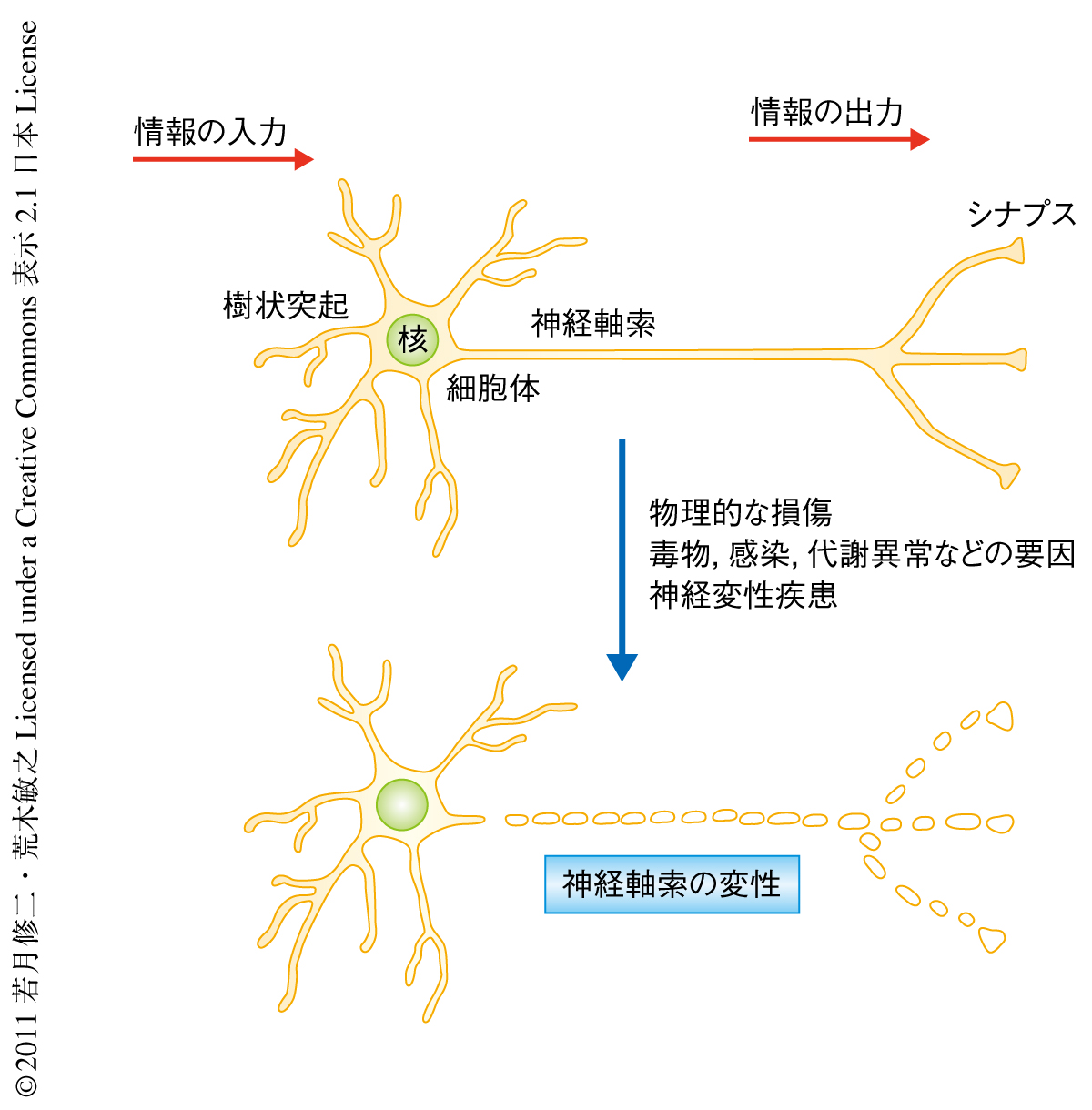
神経軸索の変性はアルツハイマー病,パーキンソン病などの神経変性疾患に共通して認められる所見であり,その制御機構を明らかにすることは疾患の抑制,さらには治療方法の開発において非常に重要な意味をもつものと考えられる(図1).これまでに,軸索の変性の制御機構はニューロンの細胞体から伸びた突起構造としての軸索が崩壊する受動的な過程ではなく,細胞内での反応を介した能動的な過程であることがさまざまな報告から示唆されている1).たとえば,微小管の脱重合やニューロフィラメントの分解など,軸索を構成する細胞骨格の変性はユビキチン-プロテアソーム系により制御されていることが知られている2).ユビキチン-プロテアソーム系は選択的なタンパク質分解を介しさまざまな細胞内シグナル伝達カスケードを制御することが知られている.これらのことから,細胞骨格系の維持にかかわるタンパク質を標的とするユビキチンリガーゼが軸索の変性の進行を制御しており,そのようなユビキチンリガーゼを同定することは軸索の変性の進行機構の詳細を理解するうえできわめて重要であると考えられる.
この論文で,筆者らは,セリン-スレオニンキナーゼであるAKTがユビキチンリガーゼZNRF1(zinc and ring finger 1)を介しプロテアソームに依存的に分解されて軸索から消失することが,軸索の変性を促進することを明らかにした.軸索に存在するAKTはリン酸化を介してGSK3B(glycogen synthase kinase 3β)のキナーゼ活性を抑制するはたらきがあり,軸索の変性が開始されるとZNRF1によるユビキチン化によりAKTが分解されることでこの抑制が失われ,活性化したGSK3Bによるリン酸化を介してCRMP2(collapsin response mediator protein 2)が阻害され微小管が崩壊することが,軸索の変性を進行させるものと考えられた.今回の結果から,AKTを介したリン酸化シグナル伝達カスケードは軸索の変性を負に制御する反応経路であり,ZNRF1はプロテアソームに依存的なタンパク質分解を介してこのカスケードを制御することにより軸索の安定性や変性を制御する“鍵タンパク質”であることが示された.
リン酸化反応はタンパク質の機能を制御する主要な細胞内反応のひとつである.タンパク質のリン酸化反応が軸索の安定性や変性を制御しているものと考え,マウス胚より調製した後根神経節を培養し軸索を伸長させたのち神経節を取り除くことで細胞体から切り離された軸索が変性する過程を経時的に観察できるin vitro軸索変性モデルを用いて,キナーゼの阻害剤をくわえることで軸索の変性を遅延させる阻害剤を探索した結果,それぞれ独立した数種類のキナーゼに対する阻害剤を同定した.同定した阻害剤のうち,とくにGSK3Bに対する阻害剤は,今回の実験でおもに使用したTDZDのほか,SB21673,5-iodo-indirubin-3’-monoximeなどが重複して同定され,かつ,軸索の変性過程で観察される微小管の脱重合,ニューロフィラメントの分解,細胞膜の崩壊など,軸索の構造的な変化を幅広く抑制することがわかった.GSK3Bは微小管の重合と脱重合とを制御することにより軸索の伸長を制御することが知られている3,4).これらのことから,GSK3Bと軸索の変性との関連についてさらにくわしく調べることにした.
GSK3Bは9番目のセリン残基がリン酸化されることにより不活性型となる.この不活性型GSK3Bを特異的に検出する抗体を用いて変性過程にある軸索におけるGSK3Bの活性化状態の変化をウェスタンブロット法により経時的に調べたところ,変性の開始ののちGSK3Bの存在量はほとんど変動を示さないが,不活性型GSK3Bは徐々に減少することがわかった.GSK3BはAKTによりリン酸化されキナーゼ活性が抑制されることが知られている.変性過程にある軸索におけるAKTについて調べたところ,AKTの存在量ならびにキナーゼ活性は変性の進行にともない減少していた.このことから,軸索の変性が進行するにつれAKTが軸索から消失することにより,AKTによる活性抑制が失われてGSK3Bが活性化するものと予想された.
それでは,軸索の変性過程においてAKTはどのように軸索から消失するのだろうか? さきに述べたように,軸索の変性はユビキチン-プロテアソーム系により制御されている.これまで,AKTのユビキチン-プロテアソーム系による分解についてさまざまな細胞種で報告されており,とくに,ニューロンにおいては神経突起の形成の制御と深く関連することが報告されている5,6).プロテアソーム阻害剤により変性を抑制させた軸索におけるAKTについて調べたところ,変性の開始ののち観察されたAKTの存在量ならびにキナーゼ活性の減少が抑制されており,かつ,このときAKTはポリユビキチン化されていることがわかった.これらの結果から,軸索の変性過程においてAKTはユビキチン-プロテアソーム系により分解されることが明らかになった.
ZNRF1は筆者らが同定したユビキチンリガーゼであり,幼弱期から成熟期までのニューロンに普遍的に発現する7).筆者らは,ZNRF1の機能解析を行う過程で,ユビキチンリガーゼ活性を欠く不活性型ZNRF1変異体を過剰発現することにより軸索の変性が顕著に抑制されることに気づいていた.そこで,ZNRF1を介したプロテアソームに依存的なAKTの分解制御の可能性について,無細胞系あるいは培養細胞を用いた強制発現系などにより検討したところ,ZNRF1はAKTと直接に結合してユビキチン化し分解を促進することなどが明らかになり,ZNRF1がAKTをプロテアソームに依存的なタンパク質分解に導くことが証明された.
ZNRF1はAKTをプロテアソームに依存的なタンパク質分解に導くことから,この制御機構と軸索の変性との関連について調べた.ZNRF1,AKT,GSK3Bそれぞれについて野生型と変異体をアデノウイルスベクターにより培養後根神経節に導入し,in vitro軸索変性モデルによる軸索の変性への影響を検討した.その結果,ユビキチンリガーゼ活性を欠く不活性型ZNRF1変異体,野生型AKTおよび活性化型AKT,不活性型GSK3B変異体は軸索の変性を抑制するが,野生型ZNRF1,野生型GSK3B,活性化型GSK3B変異体は軸索の変性には影響しないことがわかった.内在性のZNRF1あるいはGSK3Bの発現をRNAi法によりノックダウンすることでも軸索の変性は抑制された.これらのことから,AKTを介したリン酸化シグナル伝達カスケードは軸索の変性を負に制御する反応経路であり,ZNRF1はプロテアソームに依存的なタンパク質分解を介しこのカスケードを制御することにより軸索の安定性や変性を制御する“鍵タンパク質”であることが明らかになった.
GSK3Bはさまざまな細胞内シグナル伝達系の制御に関与している.軸索の変性過程において活性化したGSK3Bの標的タンパク質の候補として,軸索に発現の認められるCRMP2,MAP1B(microtubule associated protein 1B),βカテニン,APC(adenomatous polyposis coli)を選んだ.これらのタンパク質はいずれも軸索の変性の進行にともない存在量が減少し変性の開始ののち24時間でほとんど検出されなくなるが,プロテアソーム阻害剤により軸索の変性を抑制するとCRMP2の減少のみが顕著に抑制されることがわかった.CMRP2はチューブリンαβヘテロ二量体と結合することにより微小管の重合を促進し安定化させるはたらきがある.このチューブリンとの結合はGSK3Bによりリン酸化に依存的な制御をうけており,CRMP2は514番目のスレオニン残基がリン酸化されるとチューブリンとの結合能を失い微小管の脱重合が促進され不安定化することが知られている4).軸索の変性過程においてこの514番目のスレオニン残基のリン酸化の状態が変動するのかどうか調べたところ,変性の開始ののち6時間でリン酸化は亢進し,かつ,このリン酸化の亢進はGSK3Bの阻害のみならずプロテアソームを阻害することでも打ち消されることがわかった.さらに,非リン酸化型CRMP2変異体をアデノウイルスベクターにより培養後根神経節に導入して軸索の変性への影響を検討したところ,その進行は顕著に抑制された.これらのことから,CRMP2は軸索の変性におけるGSK3Bの主要な標的タンパク質であることが示された.このように,軸索の変性はZNRF1-AKT-GSK3B-CRMP2シグナル伝達カスケードにより制御されているものと考えられ,このカスケードをどのステップで阻害しても軸索の変性を抑制できることが示唆された.
それでは,ZNRF1-AKT-GSK3B-CRMP2シグナル伝達カスケードの阻害によりもたらされる軸索の変性の抑制効果は動物個体のレベルでも有効なのだろうか? 視神経変性モデルを用いてこのことを検証した.ZNRF1-AKT-GSK3B-CRMP2シグナル伝達カスケードを阻害する,たとえば,不活性型ZNRF1変異体や不活性型GSK3B変異体などをアデノウイルスベクターによりマウスの眼球へ注入し網膜神経節のニューロンに強制発現させたのち,視神経を切断することにより軸索の変性を誘導した.その結果,ZNRF1-AKT-GSK3B-CRMP2シグナル伝達カスケードをどのステップで阻害しても視神経の変性を抑制できることがわかった.
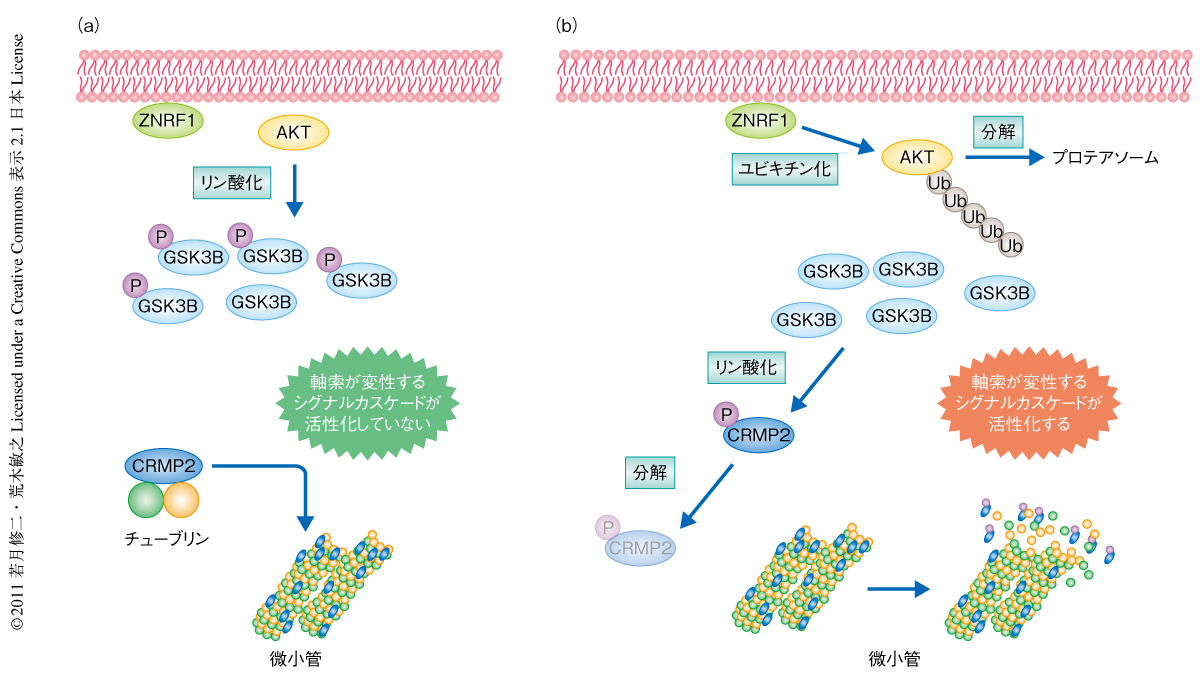
この論文で筆者らは,軸索の変性を制御するユビキチンリガーゼZNRF1を同定した.ZNRF1はAKTをプロテアソームに依存的に分解することで,GSK3Bの活性化とそれにともなうCRMP2のリン酸化を促進し微小管を不安定化させた.GSK3Bはさまざまな細胞内反応を制御することが知られているが,非リン酸化型CRMP2変異体による軸索の変性の抑制効果はプロテアソーム阻害による抑制効果と同等であったことから,CRMP2を介するこのリン酸化シグナル伝達カスケードがZNRF1により制御される主要な反応経路であると考えられた(図2).
ニューロンにおけるユビキチン-プロテアソーム系による細胞骨格の制御については,これまで,軸索の伸長の調節機構が報告されている.この例では,ユビキチンリガーゼPHR1がDLK1を分解しそのキナーゼ活性を抑制することが微小管の脱重合を促進するという8).興味深いことに,DLK1のキナーゼ活性を阻害すると軸索の変性の遅延することが報告されているが9),その効果は弱く,DLK1は軸索の変性を負に制御するというより,むしろ軸索の変性を促進するものと考えられる.これとは異なり,筆者らが明らかにしたAKTを介したリン酸化シグナル伝達カスケードは軸索の変性を負に制御する反応経路であり,その破綻は軸索の変性を促進するものと考えられた.
一方,欠失変異体を用いた結合実験により,ZNRF1はジンクフィンガー領域を介してAKTのPHドメインと結合し,また,PHドメインに存在する脊椎動物において高度に保存された5つのリシン残基がZNRF1によるユビキチン化に重要であることが明らかになった.これらのリシン残基はZNRF1とは別のユビキチンリガーゼTRAF6においてもユビキチン化の標的となることが報告されている10).TRAF6によるAKTのユビキチン化はユビキチンの63番目のリシン残基を介するK63連結型であり,AKTの細胞膜への局在化とそれにともなうキナーゼ活性の亢進を促す.これとは異なり,ZNRF1によるAKTのユビキチン化はユビキチンの48番目のリシン残基を介するK48連結型であり,AKTをプロテアソームに依存的なタンパク質分解に導くものと考えられた.このように,AKTのPHドメインにおけるユビキチン化はユビキチン連結様式の異なる2つのユビキチンリガーゼにより制御されており,それによりAKTの安定性やキナーゼ活性が調節されている可能性がある.このことは,AKTの活性の調節機構としても興味深い知見である.
今回の結果より,AKTを介したリン酸化シグナル伝達カスケードの活性化により軸索の変性を抑制できることが示された.パーキンソン病に代表されるように,多くの神経変性疾患は進行が遅く,発症ののちの早期にAKTを介したリン酸化シグナル伝達カスケードを活性化できれば軸索の変性を遅延もしくは停止でき,疾患を完全に治療できない場合にも神経の機能を維持できる期間を延長できる可能性がある.今後は,AKT活性化剤の利用,ならびに,ZNRF1によるAKTのユビキチン化を阻害する低分子化合物の探索など,AKTを介したリン酸化シグナル伝達カスケードを活性化する方法を開発することにより,軸索の保護にもとづく神経変性疾患の新たな治療戦略の創成されることが期待される.
略歴:1999年 東京大学大学院農学生命科学研究科博士課程 修了,2001年 京都大学再生医科学研究所 リサーチアソシエイト,同 助手を経て,2006年より国立精神・神経医療研究センター神経研究所 室長.
研究テーマ:末梢神経を中心に,おもに,軸索の変性および再生の分子機構,シュワン細胞の髄鞘の形成機構.
荒木 敏之(Toshiyuki Araki)
国立精神・神経医療研究センター神経研究所 部長.
研究室URL:http://www.ncnp.go.jp/nin/guide/r5/index.html
© 2011 若月修二・荒木敏之 Licensed under CC 表示 2.1 日本
(国立精神・神経医療研究センター神経研究所 疾病研究第5部)
email:若月修二,荒木敏之
DOI: 10.7875/first.author.2011.177
ZNRF1 promotes Wallerian degeneration by degrading AKT to induce GSK3B-dependent CRMP2 phosphorylation.
Shuji Wakatsuki, Fuminori Saitoh, Toshiyuki Araki
Nature Cell Biology, 13, 1415-1423 (2011)
要 約
神経軸索の変性は多くの神経変性疾患に共通する所見であり,その制御機構を明らかにすることは疾患の抑制,さらには治療方法の開発に重要な意味をもつものと考えられる.今回,筆者らは,セリン-スレオニンキナーゼであるAKTがユビキチンリガーゼZNRF1を介しプロテアソームに依存的に分解されて軸索から消失することが,軸索の変性を促進することを明らかにした.AKTにはリン酸化を介しGSK3Bを抑制するはたらきがあり,軸索の変性が開始されるとAKTの分解によりこの抑制が失われ,活性化したGSK3Bがリン酸化を介しCRMP2を不活性化することが微小管の崩壊を促進し軸索の変性を進行させるものと考えられた.これらの結果は,AKTを介したリン酸化シグナル伝達カスケードが軸索の安定性や変性を制御する主要な反応経路であることを示すとともに,この反応経路を抑制する方法を開発することが神経疾患の症状改善や進行の抑制につながる可能性を強く示唆した.
はじめに
神経軸索の変性はアルツハイマー病,パーキンソン病などの神経変性疾患に共通して認められる所見であり,その制御機構を明らかにすることは疾患の抑制,さらには治療方法の開発において非常に重要な意味をもつものと考えられる(図1).これまでに,軸索の変性の制御機構はニューロンの細胞体から伸びた突起構造としての軸索が崩壊する受動的な過程ではなく,細胞内での反応を介した能動的な過程であることがさまざまな報告から示唆されている1).たとえば,微小管の脱重合やニューロフィラメントの分解など,軸索を構成する細胞骨格の変性はユビキチン-プロテアソーム系により制御されていることが知られている2).ユビキチン-プロテアソーム系は選択的なタンパク質分解を介しさまざまな細胞内シグナル伝達カスケードを制御することが知られている.これらのことから,細胞骨格系の維持にかかわるタンパク質を標的とするユビキチンリガーゼが軸索の変性の進行を制御しており,そのようなユビキチンリガーゼを同定することは軸索の変性の進行機構の詳細を理解するうえできわめて重要であると考えられる.
この論文で,筆者らは,セリン-スレオニンキナーゼであるAKTがユビキチンリガーゼZNRF1(zinc and ring finger 1)を介しプロテアソームに依存的に分解されて軸索から消失することが,軸索の変性を促進することを明らかにした.軸索に存在するAKTはリン酸化を介してGSK3B(glycogen synthase kinase 3β)のキナーゼ活性を抑制するはたらきがあり,軸索の変性が開始されるとZNRF1によるユビキチン化によりAKTが分解されることでこの抑制が失われ,活性化したGSK3Bによるリン酸化を介してCRMP2(collapsin response mediator protein 2)が阻害され微小管が崩壊することが,軸索の変性を進行させるものと考えられた.今回の結果から,AKTを介したリン酸化シグナル伝達カスケードは軸索の変性を負に制御する反応経路であり,ZNRF1はプロテアソームに依存的なタンパク質分解を介してこのカスケードを制御することにより軸索の安定性や変性を制御する“鍵タンパク質”であることが示された.
1.GSK3Bの活性を阻害すると軸索の変性が遅延する
リン酸化反応はタンパク質の機能を制御する主要な細胞内反応のひとつである.タンパク質のリン酸化反応が軸索の安定性や変性を制御しているものと考え,マウス胚より調製した後根神経節を培養し軸索を伸長させたのち神経節を取り除くことで細胞体から切り離された軸索が変性する過程を経時的に観察できるin vitro軸索変性モデルを用いて,キナーゼの阻害剤をくわえることで軸索の変性を遅延させる阻害剤を探索した結果,それぞれ独立した数種類のキナーゼに対する阻害剤を同定した.同定した阻害剤のうち,とくにGSK3Bに対する阻害剤は,今回の実験でおもに使用したTDZDのほか,SB21673,5-iodo-indirubin-3’-monoximeなどが重複して同定され,かつ,軸索の変性過程で観察される微小管の脱重合,ニューロフィラメントの分解,細胞膜の崩壊など,軸索の構造的な変化を幅広く抑制することがわかった.GSK3Bは微小管の重合と脱重合とを制御することにより軸索の伸長を制御することが知られている3,4).これらのことから,GSK3Bと軸索の変性との関連についてさらにくわしく調べることにした.
2.軸索の変性過程においてAKTはユビキチン-プロテアソーム系により分解される
GSK3Bは9番目のセリン残基がリン酸化されることにより不活性型となる.この不活性型GSK3Bを特異的に検出する抗体を用いて変性過程にある軸索におけるGSK3Bの活性化状態の変化をウェスタンブロット法により経時的に調べたところ,変性の開始ののちGSK3Bの存在量はほとんど変動を示さないが,不活性型GSK3Bは徐々に減少することがわかった.GSK3BはAKTによりリン酸化されキナーゼ活性が抑制されることが知られている.変性過程にある軸索におけるAKTについて調べたところ,AKTの存在量ならびにキナーゼ活性は変性の進行にともない減少していた.このことから,軸索の変性が進行するにつれAKTが軸索から消失することにより,AKTによる活性抑制が失われてGSK3Bが活性化するものと予想された.
それでは,軸索の変性過程においてAKTはどのように軸索から消失するのだろうか? さきに述べたように,軸索の変性はユビキチン-プロテアソーム系により制御されている.これまで,AKTのユビキチン-プロテアソーム系による分解についてさまざまな細胞種で報告されており,とくに,ニューロンにおいては神経突起の形成の制御と深く関連することが報告されている5,6).プロテアソーム阻害剤により変性を抑制させた軸索におけるAKTについて調べたところ,変性の開始ののち観察されたAKTの存在量ならびにキナーゼ活性の減少が抑制されており,かつ,このときAKTはポリユビキチン化されていることがわかった.これらの結果から,軸索の変性過程においてAKTはユビキチン-プロテアソーム系により分解されることが明らかになった.
3.ユビキチンリガーゼZNRF1はAKTをプロテアソームに依存的なタンパク質分解に導く
ZNRF1は筆者らが同定したユビキチンリガーゼであり,幼弱期から成熟期までのニューロンに普遍的に発現する7).筆者らは,ZNRF1の機能解析を行う過程で,ユビキチンリガーゼ活性を欠く不活性型ZNRF1変異体を過剰発現することにより軸索の変性が顕著に抑制されることに気づいていた.そこで,ZNRF1を介したプロテアソームに依存的なAKTの分解制御の可能性について,無細胞系あるいは培養細胞を用いた強制発現系などにより検討したところ,ZNRF1はAKTと直接に結合してユビキチン化し分解を促進することなどが明らかになり,ZNRF1がAKTをプロテアソームに依存的なタンパク質分解に導くことが証明された.
4.ZNRF1は軸索の変性を制御する
ZNRF1はAKTをプロテアソームに依存的なタンパク質分解に導くことから,この制御機構と軸索の変性との関連について調べた.ZNRF1,AKT,GSK3Bそれぞれについて野生型と変異体をアデノウイルスベクターにより培養後根神経節に導入し,in vitro軸索変性モデルによる軸索の変性への影響を検討した.その結果,ユビキチンリガーゼ活性を欠く不活性型ZNRF1変異体,野生型AKTおよび活性化型AKT,不活性型GSK3B変異体は軸索の変性を抑制するが,野生型ZNRF1,野生型GSK3B,活性化型GSK3B変異体は軸索の変性には影響しないことがわかった.内在性のZNRF1あるいはGSK3Bの発現をRNAi法によりノックダウンすることでも軸索の変性は抑制された.これらのことから,AKTを介したリン酸化シグナル伝達カスケードは軸索の変性を負に制御する反応経路であり,ZNRF1はプロテアソームに依存的なタンパク質分解を介しこのカスケードを制御することにより軸索の安定性や変性を制御する“鍵タンパク質”であることが明らかになった.
5.GSK3BはCRMP2をリン酸化することにより不活性化し微小管の脱重合を促進する
GSK3Bはさまざまな細胞内シグナル伝達系の制御に関与している.軸索の変性過程において活性化したGSK3Bの標的タンパク質の候補として,軸索に発現の認められるCRMP2,MAP1B(microtubule associated protein 1B),βカテニン,APC(adenomatous polyposis coli)を選んだ.これらのタンパク質はいずれも軸索の変性の進行にともない存在量が減少し変性の開始ののち24時間でほとんど検出されなくなるが,プロテアソーム阻害剤により軸索の変性を抑制するとCRMP2の減少のみが顕著に抑制されることがわかった.CMRP2はチューブリンαβヘテロ二量体と結合することにより微小管の重合を促進し安定化させるはたらきがある.このチューブリンとの結合はGSK3Bによりリン酸化に依存的な制御をうけており,CRMP2は514番目のスレオニン残基がリン酸化されるとチューブリンとの結合能を失い微小管の脱重合が促進され不安定化することが知られている4).軸索の変性過程においてこの514番目のスレオニン残基のリン酸化の状態が変動するのかどうか調べたところ,変性の開始ののち6時間でリン酸化は亢進し,かつ,このリン酸化の亢進はGSK3Bの阻害のみならずプロテアソームを阻害することでも打ち消されることがわかった.さらに,非リン酸化型CRMP2変異体をアデノウイルスベクターにより培養後根神経節に導入して軸索の変性への影響を検討したところ,その進行は顕著に抑制された.これらのことから,CRMP2は軸索の変性におけるGSK3Bの主要な標的タンパク質であることが示された.このように,軸索の変性はZNRF1-AKT-GSK3B-CRMP2シグナル伝達カスケードにより制御されているものと考えられ,このカスケードをどのステップで阻害しても軸索の変性を抑制できることが示唆された.
6.ZNRF1-AKT-GSK3B-CRMP2シグナル伝達カスケードの阻害は視神経の変性を抑制する
それでは,ZNRF1-AKT-GSK3B-CRMP2シグナル伝達カスケードの阻害によりもたらされる軸索の変性の抑制効果は動物個体のレベルでも有効なのだろうか? 視神経変性モデルを用いてこのことを検証した.ZNRF1-AKT-GSK3B-CRMP2シグナル伝達カスケードを阻害する,たとえば,不活性型ZNRF1変異体や不活性型GSK3B変異体などをアデノウイルスベクターによりマウスの眼球へ注入し網膜神経節のニューロンに強制発現させたのち,視神経を切断することにより軸索の変性を誘導した.その結果,ZNRF1-AKT-GSK3B-CRMP2シグナル伝達カスケードをどのステップで阻害しても視神経の変性を抑制できることがわかった.
おわりに
この論文で筆者らは,軸索の変性を制御するユビキチンリガーゼZNRF1を同定した.ZNRF1はAKTをプロテアソームに依存的に分解することで,GSK3Bの活性化とそれにともなうCRMP2のリン酸化を促進し微小管を不安定化させた.GSK3Bはさまざまな細胞内反応を制御することが知られているが,非リン酸化型CRMP2変異体による軸索の変性の抑制効果はプロテアソーム阻害による抑制効果と同等であったことから,CRMP2を介するこのリン酸化シグナル伝達カスケードがZNRF1により制御される主要な反応経路であると考えられた(図2).
ニューロンにおけるユビキチン-プロテアソーム系による細胞骨格の制御については,これまで,軸索の伸長の調節機構が報告されている.この例では,ユビキチンリガーゼPHR1がDLK1を分解しそのキナーゼ活性を抑制することが微小管の脱重合を促進するという8).興味深いことに,DLK1のキナーゼ活性を阻害すると軸索の変性の遅延することが報告されているが9),その効果は弱く,DLK1は軸索の変性を負に制御するというより,むしろ軸索の変性を促進するものと考えられる.これとは異なり,筆者らが明らかにしたAKTを介したリン酸化シグナル伝達カスケードは軸索の変性を負に制御する反応経路であり,その破綻は軸索の変性を促進するものと考えられた.
一方,欠失変異体を用いた結合実験により,ZNRF1はジンクフィンガー領域を介してAKTのPHドメインと結合し,また,PHドメインに存在する脊椎動物において高度に保存された5つのリシン残基がZNRF1によるユビキチン化に重要であることが明らかになった.これらのリシン残基はZNRF1とは別のユビキチンリガーゼTRAF6においてもユビキチン化の標的となることが報告されている10).TRAF6によるAKTのユビキチン化はユビキチンの63番目のリシン残基を介するK63連結型であり,AKTの細胞膜への局在化とそれにともなうキナーゼ活性の亢進を促す.これとは異なり,ZNRF1によるAKTのユビキチン化はユビキチンの48番目のリシン残基を介するK48連結型であり,AKTをプロテアソームに依存的なタンパク質分解に導くものと考えられた.このように,AKTのPHドメインにおけるユビキチン化はユビキチン連結様式の異なる2つのユビキチンリガーゼにより制御されており,それによりAKTの安定性やキナーゼ活性が調節されている可能性がある.このことは,AKTの活性の調節機構としても興味深い知見である.
今回の結果より,AKTを介したリン酸化シグナル伝達カスケードの活性化により軸索の変性を抑制できることが示された.パーキンソン病に代表されるように,多くの神経変性疾患は進行が遅く,発症ののちの早期にAKTを介したリン酸化シグナル伝達カスケードを活性化できれば軸索の変性を遅延もしくは停止でき,疾患を完全に治療できない場合にも神経の機能を維持できる期間を延長できる可能性がある.今後は,AKT活性化剤の利用,ならびに,ZNRF1によるAKTのユビキチン化を阻害する低分子化合物の探索など,AKTを介したリン酸化シグナル伝達カスケードを活性化する方法を開発することにより,軸索の保護にもとづく神経変性疾患の新たな治療戦略の創成されることが期待される.
文 献
- Coleman, M. P. & Freeman, M. R.: Wallerian degeneration, wlds, and nmnat. Annu. Rev. Neurosci., 33, 245-267 (2010)[PubMed]
- Zhai, Q., Wang, J., Kim, A. et al.: Involvement of the ubiquitin-proteasome system in the early stages of wallerian degeneration. Neuron, 39, 217-225 (2003)[PubMed]
- Zhou, F. Q. & Snider, W. D.: Cell biology: GSK-3β and microtubule assembly in axons. Science, 308, 211-214 (2005)[PubMed]
- Yoshimura, T., Kawano, Y., Arimura, N. et al.: GSK-3β regulates phosphorylation of CRMP-2 and neuronal polarity. Cell, 120, 137-149 (2005)[PubMed]
- Yan, D., Guo, L. & Wang, Y.: Requirement of dendritic Akt degradation by the ubiquitin-proteasome system for neuronal polarity. J. Cell Biol., 174, 415-424 (2006)[PubMed]
- Riesterer, O., Zingg, D., Hummerjohann, J. et al.: Degradation of PKB/Akt protein by inhibition of the VEGF receptor/mTOR pathway in endothelial cells. Oncogene, 23, 4624-4635 (2004)[PubMed]
- Araki, T. & Milbrandt, J.: ZNRF proteins constitute a family of presynaptic E3 ubiquitin ligases. J. Neurosci., 23, 9385-9394 (2003)[PubMed]
- Lewcock, J. W., Genoud, N., Lettieri, K. et al.: The ubiquitin ligase Phr1 regulates axon outgrowth through modulation of microtubule dynamics. Neuron, 56, 604-620 (2007)[PubMed]
- Miller, B. R., Press, C., Daniels, R. W. et al.: A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat. Neurosci., 12, 387-389 (2009)[PubMed]
- Yang, W. L., Wang, J., Chan, C. H. et al.: The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science, 325, 1134-1138 (2009)[PubMed]
著者プロフィール
略歴:1999年 東京大学大学院農学生命科学研究科博士課程 修了,2001年 京都大学再生医科学研究所 リサーチアソシエイト,同 助手を経て,2006年より国立精神・神経医療研究センター神経研究所 室長.
研究テーマ:末梢神経を中心に,おもに,軸索の変性および再生の分子機構,シュワン細胞の髄鞘の形成機構.
荒木 敏之(Toshiyuki Araki)
国立精神・神経医療研究センター神経研究所 部長.
研究室URL:http://www.ncnp.go.jp/nin/guide/r5/index.html
© 2011 若月修二・荒木敏之 Licensed under CC 表示 2.1 日本
