膵臓β細胞における糖鎖修飾とグルコースの輸送低下を介する糖尿病の発症経路
大坪和明1・Jamey D. Marth 2
(1理化学研究所基幹研究所 ケミカルバイオロジー研究領域疾患糖鎖研究チーム,2米国Sanford-Burnham Medical Research Institute,Center for Nanomedicine)
email:大坪和明
DOI: 10.7875/first.author.2011.149
Pathway to diabetes through attenuation of pancreatic beta cell glycosylation and glucose transport.
Kazuaki Ohtsubo, Mark Z. Chen, Jerrold M. Olefsky, Jamey D. Marth
Nature Medicine, 17, 1067-1075 (2011)
これまでの糖尿病の研究において,食餌,肥満,インスリン抵抗性,インスリン分泌不全を結びつける分子機構が探求されてきた.筆者らは,高脂肪食を摂取したマウスの膵臓β細胞や遊離脂肪酸で処理した膵臓β細胞の解析から,細胞環境における遊離脂肪酸の濃度の上昇が膵臓β細胞において転写因子であるFOXA2およびHNF1αの核外への輸送と発現の減少をひき起こし,その結果,膵臓β細胞において糖鎖修飾を担当する糖転移酵素N-アセチルグルコサミニルトランスフェラーゼIVaの発現が減少し,血糖に応じたインスリン分泌機能が障害されて,さまざまなメタボリック症候群の兆候がもたらされることを明らかにした.これらの異常はヒト2型糖尿病患者に由来する膵島細胞でも観察されたことから,ヒト2型糖尿病の発症経路の重要なパートに位置するものと考えられた.一方,膵臓β細胞に特異的なN-アセチルグルコサミニルトランスフェラーゼIVaの発現により高脂肪食の負荷で誘導される膵臓β細胞の障害およびメタボリック症候群が予防されたことから,この酵素の発現および機能の維持に関与する分子が糖尿病の治療標的となる可能性が示された.
2型糖尿病は遺伝的な素因にくわえて細胞環境因子を含む複雑な要因に由来するインスリン機能不全を原因とする糖・脂質代謝異常症である1).2型糖尿病の患者はこの20年でおよそ倍増すると考えられているが,これは現代の世界的な食生活の変化および肥満と密接に関与していると考えられてきた.実際の糖尿病患者の多くは糖尿病予備軍といわれる一部の過体重あるいは肥満者から発症するわけであるが,近年,この糖尿病予備軍が急増しており新規患者の増加の源となっている2).食生活環境の変化,いわゆる,高脂肪食や西洋型の食事が肥満をひき起こし,糖尿病の発症の素因となっていることが知られているが,食餌や肥満がどのようにして2型糖尿病の病態をひき起こすのかは十分に理解されていなかった3).
インスリン機能不全は2型糖尿病の代謝的な特徴である.遺伝的なインスリン機能不全は2型糖尿病の原因としてはほんのわずかであるが,一方,インスリン分泌障害およびインスリン抵抗性の惹起は多くの2型糖尿病の病態形成に実質的に深く関与している4-6).したがって,食餌あるいは肥満を引き金に生じる膵臓β細胞からのインスリン分泌障害およびインスリン抵抗性の発生の分子機構を解明することは,2型糖尿病の発症原因を理解しその予防法をさぐるうえで重要な課題である.
膵臓β細胞の機能不全は2型糖尿病の診断基準のひとつであり,グルコース刺激によるインスリン分泌の欠失により診断される.通常,膵臓β細胞は細胞の表面に発現するグルコーストランスポーター(GLUT)により細胞にグルコースを取り込むことで血糖の上昇を認識しそれに応じたインスリン分泌を可能にしている.事実,これまでのヒトやげっ歯類の糖尿病個体の解析から,グルコース刺激によるインスリン分泌の欠失が膵臓β細胞におけるグルコーストランスポーターの発現障害と関連することが報告されており,このグルコース刺激によるインスリン分泌の障害がヒト2型糖尿病の発症過程に深く関与するとされてきた7-10).
筆者らは,タンパク質に付加するN-結合型糖鎖の分岐鎖の形成にかかわる糖転移酵素N-アセチルグルコサミニルトランスフェラーゼIVa(GnT-IVa,遺伝子名はMgat4a)を欠損したマウスが2型糖尿病を発症することを見い出していた11).その糖尿病の発症機構における糖鎖機能の解析から,膵臓β細胞においてGnT-IVaはGLUT2の糖鎖修飾を行い,GLUT2はその糖鎖を介して内在性のレクチンであるガレクチンと結合し,膵臓β細胞の表面に安定的に発現していることが明らかになった.したがって,GnT-IVaによるGLUT2の糖鎖修飾は血糖に応じたグルコースの取り込みに不可欠であり,グルコース刺激によるインスリン分泌を可能にするキープレーヤーである11)(図1).
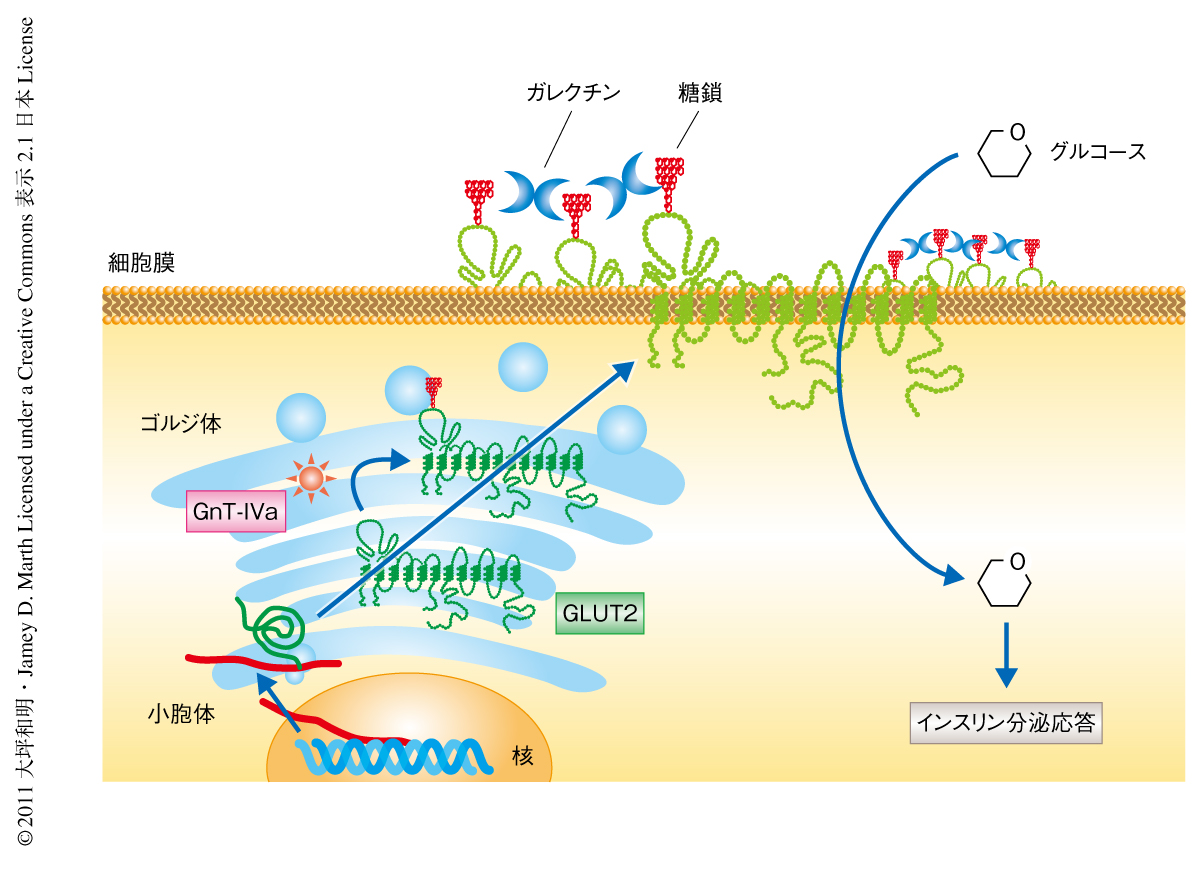
一方,野生型マウスに対し長期間にわたり高脂肪食を負荷すると,膵臓β細胞からのグルコース刺激によるインスリン分泌が障害されるとともにインスリン抵抗性が惹起されることが知られており,2型糖尿病の疾患モデルとして多くの研究に汎用されている10,12).この高脂肪食負荷マウスの膵臓β細胞ではGnT-IVaおよびGLUT2の発現が著しく障害され,GLUT2が細胞に貯留していることが明らかになった11).これらの結果は,GnT-IVaおよびGLUT2により担われるグルコースセンサー機能の障害が糖尿病の発症機構に深く関与していることを示している.
この研究において,筆者らは,糖尿病の発症過程においてGnT-IVaの機能およびGLUTの発現を調節する因子を同定し,その障害の分子機構の解明に取り組んだ.さらに,健常者および2型糖尿病患者に由来する膵臓β細胞の解析を行い,ヒト糖尿病の発症過程におけるGnT-IVaの機能および糖鎖修飾による膵臓β細胞の機能調節の意義について解析した.
これまでの筆者らの研究から,高脂肪食の摂取によりマウスの膵臓β細胞において糖転移酵素GnT-IVaをコードするMgat4a遺伝子,および,グルコーストランスポーターGLUT2をコードするGlut2遺伝子の発現が低下することが判明していた11).それと符合するように,Mgat4a遺伝子プロモーターおよびGlut2遺伝子プロモーターの活性化(ヒストンのアセチル化)のレベルが低下していた.このことから,Mgat4a遺伝子とGlut2遺伝子は同様の転写制御をうけているものと推測された.この2つの遺伝子プロモーターにおける転写因子結合モチーフの解析から,膵臓β細胞の機能維持に不可欠な転写因子であるFOXA2およびHNF1αの結合モチーフがみつかった.そこで,これら転写因子に対するsiRNAをマウスの膵臓β細胞に導入すると,Mgat4a遺伝子およびGlut2遺伝子の発現の相加的な低下が観察された.これらの結果から,この2つの遺伝子は少なくともFOXA2およびHNF1αによる転写制御をうけており,高脂肪食の負荷によりなんらかの制御異常の生じることが示された.そこで,この2つの遺伝子プロモーターへのこれら転写因子の結合がどのように変化するかをクロマチン免疫沈降法により解析した.その結果,高脂肪食負荷マウスの膵臓β細胞ではこれら転写因子のプロモーターへの結合が著しく低下していることが判明した.
この原因を探るため,マウスより膵臓β細胞を単離してこれら転写因子のタンパク質発現レベルを解析した結果,FOXA2は微減であったがHNF1αはおよそ50%にまで減少していた.さらに,膵臓β細胞におけるこれら転写因子の局在を解析した結果,標準食を摂取したマウスの膵臓β細胞では核に局在しているのに対し,高脂肪食を摂取したマウスの膵臓β細胞では核から輸送され細胞質に貯留していることが判明した.このように,高脂肪食の摂取により生じるなんらかの要因によりFOXA2およびHNF1αを介したMgat4a遺伝子およびGlut2遺伝子の転写活性化に障害がひき起こされることが判明した.
さらに,糖尿病の病態の形成過程にある膵臓β細胞を模倣するモデルとして,マウスより単離した膵臓β細胞の培養に対して遊離脂肪酸を添加して解析を行った.その結果,高脂肪食負荷マウスの膵臓β細胞で観察されていた結果と同様に,遊離脂肪酸を処理した膵臓β細胞でもFOXA2およびHNF1αが核外に輸送されることが観察され,これと一致してMgat4a遺伝子およびGlut2遺伝子の発現が著しく低下していた.
つぎに,ヒト膵臓β細胞における転写制御を解析するため,健常者に由来する膵島細胞を遊離脂肪酸により処理したところ,マウス膵臓β細胞と同様にFOXA2およびHNF1αの核外への輸送にともないMGAT4A遺伝子プロモーター,GLUT1遺伝子プロモーター,GLUT2遺伝子プロモーターへの結合の低下が観察された.これと一致して,これらの遺伝子の発現が低下しグルコース刺激によるインスリン分泌が障害された.以上の結果から,ヒト糖尿病の病態の形成過程において,膵臓β細胞への遊離脂肪酸の暴露が膵臓β細胞における糖転移酵素GnT-IVa(MGAT4A遺伝子にコードされる)およびグルコーストランスポーターGLUT1,GLUT2の発現を減弱させ,その結果,グルコースセンサー機能が障害されてグルコース刺激によるインスリン分泌が不全となることが示された.そこで確認のため,実際のヒト2型糖尿病患者より単離した膵臓β細胞の解析を行ったところ,高脂肪食負荷マウスの膵臓β細胞や遊離脂肪酸により処理した膵臓β細胞と同様に,FOXA2およびHNF1αが核外に輸送されており,それと一致してMGAT4A遺伝子,GLUT1遺伝子,GLUT2遺伝子の発現が著しく低下していた.これらを反映して膵臓β細胞の糖鎖修飾レベルの低下,膵臓β細胞の表面でのGLUT1およびGLUT2の発現レベルの低下が認められ,さらには,グルコースの取り込みが低下するとともにグルコース刺激によるインスリン分泌が著しく障害されていた.
これらの結果は,マウス膵臓β細胞と同様に,ヒト膵臓β細胞においても糖転移酵素GnT-IVaによるGLUTの糖鎖修飾がその機能維持に重要であることを表していた.そこで,ヒト膵臓β細胞におけるGLUT1およびGLUT2の糖鎖修飾とその機能についてさらに解析を行った.特異的な糖鎖構造を認識して結合するタンパク質であるレクチンを用いてヒト膵臓β細胞のGLUT1およびGLUT2の糖鎖構造の解析を行った結果,マウスGLUT2と同様に,レクチンのひとつであるガレクチンと結合する,ラクトサミン構造をもつ4本に分岐した糖鎖構造が存在することが示された.この結果から,ヒト膵臓β細胞においてもマウス膵臓β細胞と同様に,GLUT1およびGLUT2がこの糖鎖を介してガレクチンと結合し細胞の表面において安定的に発現および機能しているものと推測された.そこで,この糖鎖とガレクチンとの結合がグルコーストランスポーターの発現および機能に重要であるかどうかの検証を,ヒト膵臓β細胞の培養上清に合成糖鎖を添加してこの結合を競合的に阻害することで行った.その結果,添加した糖鎖の濃度に依存的に細胞の表面でのGLUT1およびGLUT2の発現レベルは低下し,それにともないグルコースの取り込みとグルコース刺激によるインスリン分泌が障害されることが観察された.以上の結果は,ヒト膵臓β細胞においてもGLUT1およびGLUT2にある糖鎖がガレクチンとの結合を介在しており,それにより細胞の表面におけるGLUT1およびGLUT2の安定な発現が可能となりグルコースセンサー機能をはたしていることが明らかとなった.
これまでの研究から,GLUT1およびGLUT2の発現低下および糖転移酵素GnT-IVaによる糖鎖修飾の障害が膵臓β細胞のグルコース刺激によるインスリン分泌の不全をひき起こし糖尿病の発症に深く関与していることが示された.この結果から“膵臓β細胞においてGnT-IVaを補えばその機能を保持することができ,その結果,糖代謝を正常域に維持できるのではないか?”というひとつの作業仮説にいきついた.この仮説の検証のため,GnT-IVaをコードするMGAT4A遺伝子を膵臓β細胞に特異的に発現するトランスジェニックマウスを作製し高脂肪食を負荷する実験を行った.高脂肪食の負荷により野生型マウスの膵臓β細胞ではGnT-IVaの発現が低下しGLUT2の糖鎖修飾が減少したが,このトランスジェニックマウスの膵臓β細胞ではねらいどおりGLUT2の糖鎖修飾が保持されていた.膵臓β細胞におけるGLUT2の局在を解析した結果,野生型マウスでは膵臓β細胞の表面でのGLUT2の発現が著しく低下しその多くが細胞に貯留していたが,膵臓β細胞特異的にGnT-IVaを発現するトランスジェニックマウスの膵臓β細胞では標準食と高脂肪食とで変わらず膵臓β細胞の表面のGLUT2の発現が維持されており細胞への貯留はほとんど観察されなかった.さらにこの結果と一致して,高脂肪食を負荷したこのトランスジェニックマウスの膵臓β細胞ではグルコース刺激によるインスリン分泌が正常に保持されており,高脂肪食を負荷したのちも長期にわたり血糖レベルが正常域に保持されていた.
ひるがえって,個体レベルでの良好な血糖の維持には膵臓β細胞からのインスリン分泌と末梢組織でのインスリン感受性の保持が重要である.高脂肪食を負荷したマウスの個体レベルでのインスリン感受性を測定したところ,野生型マウスでは著しくインスリン抵抗性が惹起されたのに対し,膵臓β細胞に特異的にGnT-IVaを発現するトランスジェニックマウスでは若干感受性が低下するが,筋肉や肝臓を含む全身の組織において非常に良好なインスリン感受性が保持されていた.これを裏づけるように,このトランスジェニックマウスでは筋肉および脂肪組織におけるインスリンシグナル伝達の要であるIRS-1およびAkt-1のリン酸化の制御が高脂肪食を負荷しても良好に維持されていた.これらの結果は,糖転移酵素GnT-IVaの高発現により膵臓β細胞の糖鎖修飾機能を補うことで,高脂肪食を負荷しても細胞の表面でのGLUT2の発現が十分に維持され,その結果,膵臓β細胞のグルコース刺激によるインスリン分泌を保持することができ,さらに全身のインスリン感受性を良好に保持することもできることを示していた.
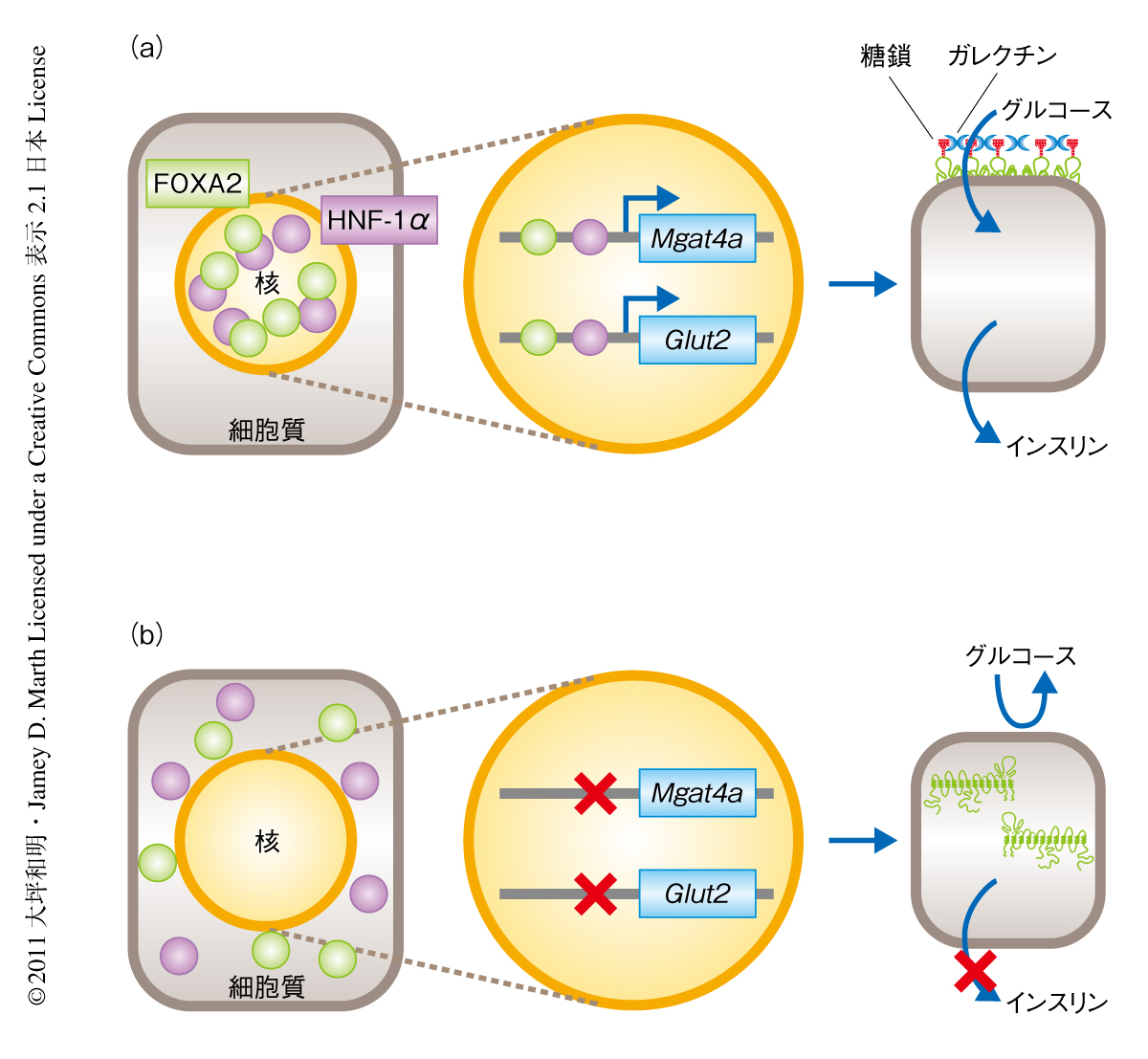
この研究により,糖尿病の発症過程における膵臓β細胞のグルコースセンサーの機能異常は,遊離脂肪酸によるMgat4a遺伝子およびGlut2遺伝子の転写異常に大きく起因することが明らかになった(図2).この発見は,複雑な要因がからみあう糖尿病の病態形成のしくみを理解するうえで非常に重要な知見である.また,糖転移酵素GnT-IVaによるGLUT2の糖鎖修飾を維持することで膵臓β細胞におけるグルコース刺激によるインスリン分泌を保持でき,さらには,個体レベルでの糖代謝の著しい改善が可能であるという知見は,糖尿病の新規の治療戦略の構築に寄与できるものと考える.今後,GnT-IVaの発現や機能の維持に作用する分子が同定されれば,ヒト2型糖尿病の予防および治療の有望な標的となる可能性がある.
略歴:2000年 京都大学大学院薬学研究科 修了,同年 米国California大学San Diego校 博士研究員および上級研究員,2008年 産業医科大学医学部 講師,2009年 大阪大学産業科学研究所 准教授を経て,2011年より理化学研究所基幹研究所 副チームリーダー.
抱負:糖鎖にコードされている生物情報を読み解き,生体や疾患機構における糖鎖機能の理解,さらには,糖鎖を用いた疾患の治療につなげていきたい.
Jamey D. Marth
米国Sanford-Burnham Medical Research InstituteにてPrincipal Investigator.
(1理化学研究所基幹研究所 ケミカルバイオロジー研究領域疾患糖鎖研究チーム,2米国Sanford-Burnham Medical Research Institute,Center for Nanomedicine)
email:大坪和明
DOI: 10.7875/first.author.2011.149
Pathway to diabetes through attenuation of pancreatic beta cell glycosylation and glucose transport.
Kazuaki Ohtsubo, Mark Z. Chen, Jerrold M. Olefsky, Jamey D. Marth
Nature Medicine, 17, 1067-1075 (2011)
この論文に出現する遺伝子・タンパク質のUniprot ID
FOXA2(P35583), HNF1α(P22361), 糖転移酵素N-アセチルグルコサミニルトランスフェラーゼIVa(Q812G0), N-アセチルグルコサミニルトランスフェラーゼIVa(Q812G0), グルコーストランスポーター, GLUT, GnT-IVa(Q812G0), Mgat4a(Q812G0), GLUT2(P14246), レクチン, ガレクチン, Glut2(P14246), 糖転移酵素GnT-IVa(Q812G0), グルコーストランスポーターGLUT2(P14246), MGAT4A(Q812G0), GLUT1(P11166), グルコーストランスポーターGLUT1(P11166), IRS-1(P35569), Akt-1(P31750), 転写因子FOXA2(P35583)
要 約
これまでの糖尿病の研究において,食餌,肥満,インスリン抵抗性,インスリン分泌不全を結びつける分子機構が探求されてきた.筆者らは,高脂肪食を摂取したマウスの膵臓β細胞や遊離脂肪酸で処理した膵臓β細胞の解析から,細胞環境における遊離脂肪酸の濃度の上昇が膵臓β細胞において転写因子であるFOXA2およびHNF1αの核外への輸送と発現の減少をひき起こし,その結果,膵臓β細胞において糖鎖修飾を担当する糖転移酵素N-アセチルグルコサミニルトランスフェラーゼIVaの発現が減少し,血糖に応じたインスリン分泌機能が障害されて,さまざまなメタボリック症候群の兆候がもたらされることを明らかにした.これらの異常はヒト2型糖尿病患者に由来する膵島細胞でも観察されたことから,ヒト2型糖尿病の発症経路の重要なパートに位置するものと考えられた.一方,膵臓β細胞に特異的なN-アセチルグルコサミニルトランスフェラーゼIVaの発現により高脂肪食の負荷で誘導される膵臓β細胞の障害およびメタボリック症候群が予防されたことから,この酵素の発現および機能の維持に関与する分子が糖尿病の治療標的となる可能性が示された.
はじめに
2型糖尿病は遺伝的な素因にくわえて細胞環境因子を含む複雑な要因に由来するインスリン機能不全を原因とする糖・脂質代謝異常症である1).2型糖尿病の患者はこの20年でおよそ倍増すると考えられているが,これは現代の世界的な食生活の変化および肥満と密接に関与していると考えられてきた.実際の糖尿病患者の多くは糖尿病予備軍といわれる一部の過体重あるいは肥満者から発症するわけであるが,近年,この糖尿病予備軍が急増しており新規患者の増加の源となっている2).食生活環境の変化,いわゆる,高脂肪食や西洋型の食事が肥満をひき起こし,糖尿病の発症の素因となっていることが知られているが,食餌や肥満がどのようにして2型糖尿病の病態をひき起こすのかは十分に理解されていなかった3).
インスリン機能不全は2型糖尿病の代謝的な特徴である.遺伝的なインスリン機能不全は2型糖尿病の原因としてはほんのわずかであるが,一方,インスリン分泌障害およびインスリン抵抗性の惹起は多くの2型糖尿病の病態形成に実質的に深く関与している4-6).したがって,食餌あるいは肥満を引き金に生じる膵臓β細胞からのインスリン分泌障害およびインスリン抵抗性の発生の分子機構を解明することは,2型糖尿病の発症原因を理解しその予防法をさぐるうえで重要な課題である.
膵臓β細胞の機能不全は2型糖尿病の診断基準のひとつであり,グルコース刺激によるインスリン分泌の欠失により診断される.通常,膵臓β細胞は細胞の表面に発現するグルコーストランスポーター(GLUT)により細胞にグルコースを取り込むことで血糖の上昇を認識しそれに応じたインスリン分泌を可能にしている.事実,これまでのヒトやげっ歯類の糖尿病個体の解析から,グルコース刺激によるインスリン分泌の欠失が膵臓β細胞におけるグルコーストランスポーターの発現障害と関連することが報告されており,このグルコース刺激によるインスリン分泌の障害がヒト2型糖尿病の発症過程に深く関与するとされてきた7-10).
筆者らは,タンパク質に付加するN-結合型糖鎖の分岐鎖の形成にかかわる糖転移酵素N-アセチルグルコサミニルトランスフェラーゼIVa(GnT-IVa,遺伝子名はMgat4a)を欠損したマウスが2型糖尿病を発症することを見い出していた11).その糖尿病の発症機構における糖鎖機能の解析から,膵臓β細胞においてGnT-IVaはGLUT2の糖鎖修飾を行い,GLUT2はその糖鎖を介して内在性のレクチンであるガレクチンと結合し,膵臓β細胞の表面に安定的に発現していることが明らかになった.したがって,GnT-IVaによるGLUT2の糖鎖修飾は血糖に応じたグルコースの取り込みに不可欠であり,グルコース刺激によるインスリン分泌を可能にするキープレーヤーである11)(図1).
一方,野生型マウスに対し長期間にわたり高脂肪食を負荷すると,膵臓β細胞からのグルコース刺激によるインスリン分泌が障害されるとともにインスリン抵抗性が惹起されることが知られており,2型糖尿病の疾患モデルとして多くの研究に汎用されている10,12).この高脂肪食負荷マウスの膵臓β細胞ではGnT-IVaおよびGLUT2の発現が著しく障害され,GLUT2が細胞に貯留していることが明らかになった11).これらの結果は,GnT-IVaおよびGLUT2により担われるグルコースセンサー機能の障害が糖尿病の発症機構に深く関与していることを示している.
この研究において,筆者らは,糖尿病の発症過程においてGnT-IVaの機能およびGLUTの発現を調節する因子を同定し,その障害の分子機構の解明に取り組んだ.さらに,健常者および2型糖尿病患者に由来する膵臓β細胞の解析を行い,ヒト糖尿病の発症過程におけるGnT-IVaの機能および糖鎖修飾による膵臓β細胞の機能調節の意義について解析した.
1.糖尿病の病態の形成過程におけるMgat4a遺伝子およびGlut2遺伝子の発現調節
これまでの筆者らの研究から,高脂肪食の摂取によりマウスの膵臓β細胞において糖転移酵素GnT-IVaをコードするMgat4a遺伝子,および,グルコーストランスポーターGLUT2をコードするGlut2遺伝子の発現が低下することが判明していた11).それと符合するように,Mgat4a遺伝子プロモーターおよびGlut2遺伝子プロモーターの活性化(ヒストンのアセチル化)のレベルが低下していた.このことから,Mgat4a遺伝子とGlut2遺伝子は同様の転写制御をうけているものと推測された.この2つの遺伝子プロモーターにおける転写因子結合モチーフの解析から,膵臓β細胞の機能維持に不可欠な転写因子であるFOXA2およびHNF1αの結合モチーフがみつかった.そこで,これら転写因子に対するsiRNAをマウスの膵臓β細胞に導入すると,Mgat4a遺伝子およびGlut2遺伝子の発現の相加的な低下が観察された.これらの結果から,この2つの遺伝子は少なくともFOXA2およびHNF1αによる転写制御をうけており,高脂肪食の負荷によりなんらかの制御異常の生じることが示された.そこで,この2つの遺伝子プロモーターへのこれら転写因子の結合がどのように変化するかをクロマチン免疫沈降法により解析した.その結果,高脂肪食負荷マウスの膵臓β細胞ではこれら転写因子のプロモーターへの結合が著しく低下していることが判明した.
この原因を探るため,マウスより膵臓β細胞を単離してこれら転写因子のタンパク質発現レベルを解析した結果,FOXA2は微減であったがHNF1αはおよそ50%にまで減少していた.さらに,膵臓β細胞におけるこれら転写因子の局在を解析した結果,標準食を摂取したマウスの膵臓β細胞では核に局在しているのに対し,高脂肪食を摂取したマウスの膵臓β細胞では核から輸送され細胞質に貯留していることが判明した.このように,高脂肪食の摂取により生じるなんらかの要因によりFOXA2およびHNF1αを介したMgat4a遺伝子およびGlut2遺伝子の転写活性化に障害がひき起こされることが判明した.
さらに,糖尿病の病態の形成過程にある膵臓β細胞を模倣するモデルとして,マウスより単離した膵臓β細胞の培養に対して遊離脂肪酸を添加して解析を行った.その結果,高脂肪食負荷マウスの膵臓β細胞で観察されていた結果と同様に,遊離脂肪酸を処理した膵臓β細胞でもFOXA2およびHNF1αが核外に輸送されることが観察され,これと一致してMgat4a遺伝子およびGlut2遺伝子の発現が著しく低下していた.
つぎに,ヒト膵臓β細胞における転写制御を解析するため,健常者に由来する膵島細胞を遊離脂肪酸により処理したところ,マウス膵臓β細胞と同様にFOXA2およびHNF1αの核外への輸送にともないMGAT4A遺伝子プロモーター,GLUT1遺伝子プロモーター,GLUT2遺伝子プロモーターへの結合の低下が観察された.これと一致して,これらの遺伝子の発現が低下しグルコース刺激によるインスリン分泌が障害された.以上の結果から,ヒト糖尿病の病態の形成過程において,膵臓β細胞への遊離脂肪酸の暴露が膵臓β細胞における糖転移酵素GnT-IVa(MGAT4A遺伝子にコードされる)およびグルコーストランスポーターGLUT1,GLUT2の発現を減弱させ,その結果,グルコースセンサー機能が障害されてグルコース刺激によるインスリン分泌が不全となることが示された.そこで確認のため,実際のヒト2型糖尿病患者より単離した膵臓β細胞の解析を行ったところ,高脂肪食負荷マウスの膵臓β細胞や遊離脂肪酸により処理した膵臓β細胞と同様に,FOXA2およびHNF1αが核外に輸送されており,それと一致してMGAT4A遺伝子,GLUT1遺伝子,GLUT2遺伝子の発現が著しく低下していた.これらを反映して膵臓β細胞の糖鎖修飾レベルの低下,膵臓β細胞の表面でのGLUT1およびGLUT2の発現レベルの低下が認められ,さらには,グルコースの取り込みが低下するとともにグルコース刺激によるインスリン分泌が著しく障害されていた.
これらの結果は,マウス膵臓β細胞と同様に,ヒト膵臓β細胞においても糖転移酵素GnT-IVaによるGLUTの糖鎖修飾がその機能維持に重要であることを表していた.そこで,ヒト膵臓β細胞におけるGLUT1およびGLUT2の糖鎖修飾とその機能についてさらに解析を行った.特異的な糖鎖構造を認識して結合するタンパク質であるレクチンを用いてヒト膵臓β細胞のGLUT1およびGLUT2の糖鎖構造の解析を行った結果,マウスGLUT2と同様に,レクチンのひとつであるガレクチンと結合する,ラクトサミン構造をもつ4本に分岐した糖鎖構造が存在することが示された.この結果から,ヒト膵臓β細胞においてもマウス膵臓β細胞と同様に,GLUT1およびGLUT2がこの糖鎖を介してガレクチンと結合し細胞の表面において安定的に発現および機能しているものと推測された.そこで,この糖鎖とガレクチンとの結合がグルコーストランスポーターの発現および機能に重要であるかどうかの検証を,ヒト膵臓β細胞の培養上清に合成糖鎖を添加してこの結合を競合的に阻害することで行った.その結果,添加した糖鎖の濃度に依存的に細胞の表面でのGLUT1およびGLUT2の発現レベルは低下し,それにともないグルコースの取り込みとグルコース刺激によるインスリン分泌が障害されることが観察された.以上の結果は,ヒト膵臓β細胞においてもGLUT1およびGLUT2にある糖鎖がガレクチンとの結合を介在しており,それにより細胞の表面におけるGLUT1およびGLUT2の安定な発現が可能となりグルコースセンサー機能をはたしていることが明らかとなった.
2.糖転移酵素GnT-IVaに依存的な糖鎖修飾による膵臓β細胞の機能維持と糖代謝の制御
これまでの研究から,GLUT1およびGLUT2の発現低下および糖転移酵素GnT-IVaによる糖鎖修飾の障害が膵臓β細胞のグルコース刺激によるインスリン分泌の不全をひき起こし糖尿病の発症に深く関与していることが示された.この結果から“膵臓β細胞においてGnT-IVaを補えばその機能を保持することができ,その結果,糖代謝を正常域に維持できるのではないか?”というひとつの作業仮説にいきついた.この仮説の検証のため,GnT-IVaをコードするMGAT4A遺伝子を膵臓β細胞に特異的に発現するトランスジェニックマウスを作製し高脂肪食を負荷する実験を行った.高脂肪食の負荷により野生型マウスの膵臓β細胞ではGnT-IVaの発現が低下しGLUT2の糖鎖修飾が減少したが,このトランスジェニックマウスの膵臓β細胞ではねらいどおりGLUT2の糖鎖修飾が保持されていた.膵臓β細胞におけるGLUT2の局在を解析した結果,野生型マウスでは膵臓β細胞の表面でのGLUT2の発現が著しく低下しその多くが細胞に貯留していたが,膵臓β細胞特異的にGnT-IVaを発現するトランスジェニックマウスの膵臓β細胞では標準食と高脂肪食とで変わらず膵臓β細胞の表面のGLUT2の発現が維持されており細胞への貯留はほとんど観察されなかった.さらにこの結果と一致して,高脂肪食を負荷したこのトランスジェニックマウスの膵臓β細胞ではグルコース刺激によるインスリン分泌が正常に保持されており,高脂肪食を負荷したのちも長期にわたり血糖レベルが正常域に保持されていた.
ひるがえって,個体レベルでの良好な血糖の維持には膵臓β細胞からのインスリン分泌と末梢組織でのインスリン感受性の保持が重要である.高脂肪食を負荷したマウスの個体レベルでのインスリン感受性を測定したところ,野生型マウスでは著しくインスリン抵抗性が惹起されたのに対し,膵臓β細胞に特異的にGnT-IVaを発現するトランスジェニックマウスでは若干感受性が低下するが,筋肉や肝臓を含む全身の組織において非常に良好なインスリン感受性が保持されていた.これを裏づけるように,このトランスジェニックマウスでは筋肉および脂肪組織におけるインスリンシグナル伝達の要であるIRS-1およびAkt-1のリン酸化の制御が高脂肪食を負荷しても良好に維持されていた.これらの結果は,糖転移酵素GnT-IVaの高発現により膵臓β細胞の糖鎖修飾機能を補うことで,高脂肪食を負荷しても細胞の表面でのGLUT2の発現が十分に維持され,その結果,膵臓β細胞のグルコース刺激によるインスリン分泌を保持することができ,さらに全身のインスリン感受性を良好に保持することもできることを示していた.
おわりに
この研究により,糖尿病の発症過程における膵臓β細胞のグルコースセンサーの機能異常は,遊離脂肪酸によるMgat4a遺伝子およびGlut2遺伝子の転写異常に大きく起因することが明らかになった(図2).この発見は,複雑な要因がからみあう糖尿病の病態形成のしくみを理解するうえで非常に重要な知見である.また,糖転移酵素GnT-IVaによるGLUT2の糖鎖修飾を維持することで膵臓β細胞におけるグルコース刺激によるインスリン分泌を保持でき,さらには,個体レベルでの糖代謝の著しい改善が可能であるという知見は,糖尿病の新規の治療戦略の構築に寄与できるものと考える.今後,GnT-IVaの発現や機能の維持に作用する分子が同定されれば,ヒト2型糖尿病の予防および治療の有望な標的となる可能性がある.
文 献
- Saltiel, A. R. & Kahn, C. R.: Insulin signaling and the regulation of glucose and lipid metabolism. Nature, 414, 799-806 (2001)[PubMed]
- Korner, J., Woods, S. C. & Woodworth, K. A.: Regulation of energy homeostasis and health consequences in obesity. Am. J. Med., 122, S12-S18 (2009)[PubMed]
- Parillo, M. & Ricardi, G.: Diet composition and the risk of type 2 diabetes: epidemiological and clinical evidence. Br. J. Nutr., 92, 7-19 (2004)[PubMed]
- Kahn, S. E.: The relative contribution of insulin resistance and beta-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia, 46, 3-19 (2003)[PubMed]
- Schenk, S., Saberi, M. & Olefsky, J. M.: Insulin sensitivity: modulation by nutrients and inflammation. J. Clin. Invest., 118, 2992-3002 (2008)[PubMed]
- Reaven, G. M.: The insulin resistance syndrome: definition and dietary approaches to treatment. Annu. Rev. Nutr., 25, 391-406 (2005)[PubMed]
- Valera, A., Solanes, G., Fernandez-Alvarez, J. et al.: Expression of GLUT-2 antisense RNA in β cells of transgenic mice leads to diabetes. J. Biol. Chem., 269, 28543-28546 (1994)[PubMed]
- Guillam, M. T., Hummler, E., Schaerer, E. et al.: Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat. Genet., 17, 327-330 (1997)[PubMed]
- Guillam, M. T., Dupraz, P. & Thorens, B.: Glucose uptake, utilization, and signaling in GLUT2-null islets. Diabetes, 49, 1485-1491 (2000)[PubMed]
- Reimer, M. K. & Ahren, B.: Altered β-cell distribution of pdx-1 and GLUT-2 after a short-term challenge with a high-fat diet in C57/BL/6J mice. Diabetes, 51, S138-S143 (2002)[PubMed]
- Ohtsubo, K., Takamatsu, S., Minowa, M. T. et al.: Dietary and genetic control of glucose transporter 2 glycosylation promotes insulin secretion in suppressing diabetes. Cell, 123, 1307-1321 (2005)[PubMed]
- Winzell, M. S. & Ahren, B.: The high-fat diet-fed mouse: a model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes, 53(suppl. 3), S215-S219 (2004)[PubMed]
著者プロフィール
略歴:2000年 京都大学大学院薬学研究科 修了,同年 米国California大学San Diego校 博士研究員および上級研究員,2008年 産業医科大学医学部 講師,2009年 大阪大学産業科学研究所 准教授を経て,2011年より理化学研究所基幹研究所 副チームリーダー.
抱負:糖鎖にコードされている生物情報を読み解き,生体や疾患機構における糖鎖機能の理解,さらには,糖鎖を用いた疾患の治療につなげていきたい.
Jamey D. Marth
米国Sanford-Burnham Medical Research InstituteにてPrincipal Investigator.
© 2011 大坪和明・Jamey D. Marth Licensed under CC 表示 2.1 日本
